










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versão impressa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.70 no.4 México Jul./Ago. 2013
CASO CLÍNICO PATOLÓGICO
Adolescente de 14 años con meningitis crónica secundaria a granulomatosis de Wegener en sistema nervioso central
Chronic meningitis secondary to Wegener's granulomatosis in the central nervous system in a 14-year-old patient
Rómulo Erick Rosales-Uribe,1 Aarón Pacheco-Ríos,2 Marco Antonio Sarmiento,3 María Argelia Escobar-Sánchez4
1 Subdirección de Atención Integral al Paciente
2 Subdirección de Enseñanza
3 Departamento de Radiología
4 Departamento de Patología
Hospital Infantil de México Federico Gómez
México D.F., México
Autor de correspondencia:
Dra. María Argelia Escobar Sánchez
Correo electrónico: bcnhim20091@hotmail.com
Fecha de recepción: 08-04-13
Fecha de aceptación: 08-05-13
Resumen de la historia clínica (A-11-44)
Se presenta la historia de un adolescente que acude trasladado de un hospital segundo nivel por cefalea y alteración en el estado de alerta.
Antecedentes heredofamiliares. Procedente de familia desintegrada, padre fuera del núcleo familiar, aparentemente sano. Vive con su madre y con hermanos de 22, 18 y 16 años. Antecedentes en ambas ramas de diabetes mellitus e hipertensión arterial sistémica.
Antecedentes no patológicos. Originario y residente del Estado de México, en casa prestada con todos los servicios. Desarrollo psicomotor normal. Inmunizaciones completas.
Antecedentes perinatales y patológicos. Producto de gesta IV de embarazo normoevolutivo atendido en centro de salud. Parto vía vaginal, peso de 3,330 g, talla no recuerda. Varicela a los 3 años. Ingirió cáustico por intento de suicidio en octubre del 2009, resultando una lesión esofágica severa, por lo que en ese ingreso se realizó gastrostomía. Transfusionales positivos en esa ocasión. Catorce días previos al ingreso del padecimiento actual presentó rinorrea hialina sin tos ni fiebre que cedió sin tratamiento.
Padecimiento actual. Inició un día antes de su ingreso al Hospital Infantil de México Federico Gómez (HIMFG). Fue llevado a otro hospital con Glasgow de 14, disartria y tos productiva. Se realizó punción lumbar por probable neuroinfección. Se obtuvo líquido cefalorraquídeo (LCR) claro, proteínas de 30 mg/dl, glucosa 70 mg/dl, eritrocitos 20 mm3, no se observaron bacterias. Biometría hemática con leucocitos 9,000/mm3, neutrófilos 67%, linfocitos 20%, hemoglobina 14.6 g/dl. Se envió al HIMFG para descartar absceso cerebral. A su ingreso se obtuvo una historia de 10 días de evolución con cefalea de inicio súbito, punzante universal. Somnolencia de 4 días de evolución progresiva, con alteración de los ciclos de sueño con hasta 15 horas de sueño y posteriormente bradilalia y bradipsiquia.
A la exploración física se encontró peso de 44 kg, talla de 1.66 m, frecuencia cardiaca (FC) 64/min, frecuencia respiratoria (FR) 14/min, TA 100/54 mmHg, temperatura 37°C.
Paciente masculino de 14 años de edad, edad aparente a la cronológica, con bradilalia y bradipsiquia, sin alteraciones en pares craneales, con tono muscular normal así como trofismo disminuido. Pupilas isocóricas normoreflécticas. Cuello normal. Campos pulmonares limpios y bien ventilados. Ruidos cardiacos normales. Abdomen con presencia de gastrostomía funcional, blando, depresible, sin dolor. Llenado capilar de 2 seg en extremidades. Glasgow 15/15 con fuerza de miembro torácico y pélvico izquierdo 4/5, reflejo osteotendinoso (ROT) miembro torácico y pélvico izquierdo +++/++++, Babinski izquierdo, marcha atáxica, sensibilidad conservada, no dismetrias ni disdiadococinesias. Kernig y Brudzinski presentes.
Presentación del caso
Subdirección de Atención Integral al Paciente (Dr. Rómulo Erick Rosales Uribe)
El caso que se analiza es el de un paciente adolescente de 14 años, con antecedentes de intento de suicidio por ingesta de cáustico, por lo que se le realizó una transposición de colon. Dos años después acudió por datos de una evolución crónica en sistema nervioso central, que ameritó un estudio multidisciplinario ante los datos de enfermedad grave en sistema nervioso central. La primera posibilidad etiológica en nuestro medio era una meningitis tuberculosa, que fue descartada en la autopsia, y se estableció el diagnóstico de granulomatosis de Wegener sin componente renal o respiratorio, lo cual es un evento muy raro.
Imagenología (Dr. Marco Antonio Sarmiento)
La radiografía de tórax inicialmente se encuentra normal a su ingreso. Evolucionó a un derrame pleural que ocupó todo el hemitórax izquierdo, para ser posteriormente bilateral.
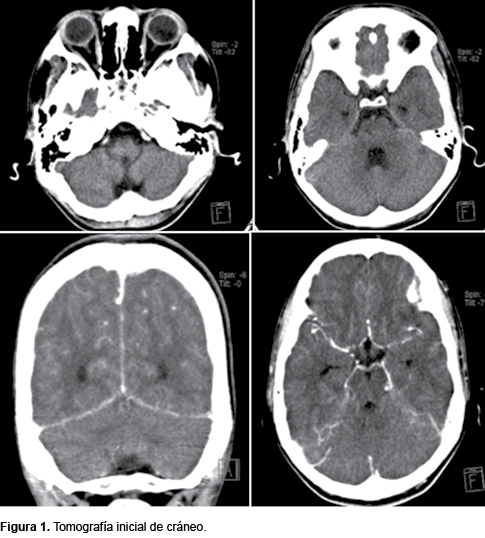
La tomografía inicial de cráneo con asimetría en puente e hipodensidad a nivel mesencefálico con un sistema ventricular normal. Al usar contraste se observó una imagen hiperdensa y reforzamiento de meninges en la tienda, con una relación de sustancia gris y blanca adecuada. El polígono de Willis se reporta normal (Figura 1). Catorce días después de realiza una nueva TAC encontrando polígono de Willis irregular, mayor reforzamiento meníngeo e hidrocefalia (Figura 2). Un mes después de su ingreso se observó la pérdida de la relación de sustancia gris y blanca, con borramiento de circunvoluciones frontales y parietales, reforzamiento meníngeo, así como múltiples áreas hipodensas con disminución de la perfusión cerebral.

En la resonancia magnética de cráneo con fase FLAIR se observó en puente una imagen hipointensa derecha asimétrica y el ensanchamiento supratentorial del sistema ventricular. En otras fases, una imagen hiperintensa asimétrica en puente. El espacio subaracnoideo hacia la convexidad se apreció de características normales. Adecuada configuración del sistema ventricular supra e infratentorial. Adecuada relación de la sustancia gris con la sustancia blanca. En el tallo cerebral se apreció una imagen amorfa hipodensa localizada en el pedúnculo cerebral derecho. El cerebelo se apreció de aspecto normal. Posterior a la inyección de medio de contraste no se observaron áreas de reforzamiento patológico. Adecuada configuración del polígono de Willis.
Dilatación del sistema ventricular supra e infratentorial ya evidente en el estudio previo. Con la inyección del medio de contraste se apreció la adecuada conformación del polígono, con aspecto arrosariado en las arterias cerebrales medias y anteriores. En el parénquima cerebral no se observaron alteraciones (Figura 2).
Se observaron cambios post quirúrgicos caracterizados por colocación de válvula de derivación en el ventrículo lateral derecho. Sin cambios en la amplitud del sistema ventricular (Figura 3).

En la radiografía de tórax se apreció vascularidad pulmonar aumentada. En el hemitórax izquierdo se observó un patrón en vidrio deslustrado. Pérdida del ángulo costo diafragmático izquierdo en relación con derrame. La silueta cardiaca de aspecto normal. En la tráquea se observó sonda orotraqueal en buena posición (Figura 4).

Cambios post quirúrgicos por colación de válvulas de derivación en ambos ventrículos laterales con adecuado funcionamiento. Pérdida de la relación de la sustancia gris de la blanca profunda con áreas de hipo perfusión vascular (Figura 5).

Diagnósticos por imagen:
• Transposición colónica
• Área de encéfalo malacia en el tallo cerebral
• Infiltrados alveolares basales de forma difusa
• Derrame pleural
• Hidrocefalia
• Datos sugestivos vasculitis en el polígono Willis
Neurología (Dr. Rubén Espinoza Montero) ¿Algún dato de aracnoiditis basal?
Imagenología (Dr. Marco Antonio Sarmiento)
Solo se observó en tentorio y la presencia de hidrocefalia.
Subdirección de Pediatría Ambulatoria (Dr. Edgar Bustos)
¿En tallo cerebral las áreas que se ven son de una mielinolisis pontina?
Imagenología (Dr. Marco Antonio Sarmiento)
No. En los casos de mielinolisis pontina se ven imágenes hiperdensas difusas en tallo cerebral y patrones de restricción en secuencia de difusión.
Coordinador (Dr. Rómulo Erick Rosales)
En la práctica clínica el resultado de una biopsia transoperatoria se tiene al momento así que, para seguir con el ejercicio clínico y que sea de ayuda para el discusor, le pido a la Dra. Argelia Escobar Sánchez que dé un reporte de esta biopsia antes de dar el reporte de la autopsia.
Patología (Dra. Argelia Escobar Sánchez)
Se recibió una biopsia transoperatoria de cerebro. Se realizaron cortes en el tejido congelado en los que se encuentra la sustancia blanca y la corteza cerebral con necrosis extensa y granulomas. Posteriormente, en el tejido fijado se realizaron múltiples tinciones buscando bacilos ácido-alcohol resistentes (BAAR), que no se encontraron. Únicamente se reportó meningoencefalitis granulomatosa, pero se sugiere en el reporte realizar reacción en cadena de la polimerasa (PCR) para tuberculosis.
Discusión
Subdirección de Enseñanza (Dr. Aarón Pacheco Ríos)
Se trató de un paciente masculino de 14 años, proveniente de un medio socioeconómico bajo. Llama la atención el antecedente de intento de suicidio a través de la ingesta de cáusticos, dado que el suicidio es una causa importante de muerte en la adolescencia (segundo lugar). Se considera que faltó un análisis a profundidad de las causas que rodearon este intento, pues era urgente identificar y controlar un nuevo intento. Posteriormente, requirió una transposición colónica y dilataciones esofágicas que hicieron sospechar que un absceso cerebral podía ser la causa de su patología, pero se descartó esta posibilidad.
Con los datos clínicos se integran los siguientes síndromes:
1. Síndrome de neurona motora superior. Caracterizado por hemiparesia, hiperreflexia y signo de Babinsky izquierdos, que indican una afección de la vía piramidal del lado derecho por debajo de la decusación de las pirámides. Llama la atención la bradilalia y bradipsiquia y, considerando los hallazgos en imagen así como el que la vía corticoespinal y vía corticobulbar dan al hombre moderno las principales características que son la destreza manual y la destreza del lenguaje, se propone que se afectaron estas dos vías a nivel de la lesión hipodensa que se observa en estudios de imagen. Esto explica la incapacidad para articular palabra, más que bradilalia o bradipsiquia.
2. Síndrome meníngeo. Al ingreso presentó irritación meníngea con signos de Kernig y Brudzinsky, más alteraciones en el líquido cefalorraquídeo, que lo catalogan como anormal por la presencia de hipoglucorraquia, hiperproteinorraquia, pleocitosis con predominio de PMN (polimorfonucleares) en 80%.
3. Meningitis crónica. Se presentó de una a cuatro semanas desde el inicio de los síntomas y las causas se dividen en infecciosas y no infecciosas. Las causas no infecciosas, como angeítis granulomatosa y enfermedad de Behçet, no parecen corresponder ante la ausencia de datos clínicos. Tampoco resultan frecuentes a los 14 años.
4. Tuberculosis meníngea. La principal causa infecciosa de meningitis crónica es la tuberculosis meníngea, que se puede apoyar con base en los hallazgos radiológicos de vasculitis, arrosariamiento del polígono de Willis más la hidrocefalia que refleja un bloqueo de la circulación de LCR. La tuberculosis meníngea en el adolescente no es sistémica;, más bien es por descarga de un foco antiguo de tuberculosis al espacio subaracnoideo. En los primeros 10 días pueden encontrarse PMN, y el cambio en los primeros días de nuestro paciente, con un incremento en proteínas en LCR y disminución de la glucosa, así como un predominio de mononucleares, apoyan el diagnóstico a pesar de tener en las tinciones de LCR negatividad a BAAR (solo en 10 a 20% de las veces es positivo, aún a pesar de una verdadera tuberculosis meníngea).
5. Síndrome de secreción inapropiada de hormona antidiurética. La presencia de hiponatremia euvolémica con sodio sérico bajo más hipotonicidad y con valores osmolares séricos de 240 mOsm con una osmolaridad urinaria elevada apoyan este diagnóstico. Sin embargo, faltaron estudios para descartar otras posibilidades, como el síndrome cerebral perdedor de sal.
6. Herniación de amígdalas cerebelosas. Ante una evolución inadecuada con un deterioro neurológico rostrocaudal más los hallazgos en TAC de una escasa diferenciación de sustancia gris y blanca, colapso ventricular y cisternas perimesencefálicas borradas hacen suponer un edema cerebral grave con una probable herniación de las amígdalas cerebelosas que lo llevaron a la muerte.
7. Descartar la posibilidad de mielinolisis pontina. Esta se presenta habitualmente varios días después de una corrección de hiponatremia crónica. El paciente presentó irritabilidad que evolucionó a agresividad, lo que pueden ser datos iniciales de una mielinolisis. Los síntomas clásicos, como cuadriparesia espástica y la parálisis pseudobulbar, reflejan el daño en los tractos corticoespinal y corticobulbar.
Es necesario evaluar la hiponatremia sintomática aguda, pues en caso de un evento agudo con síntomas no hay efectos deletéreos al corregirse, pero en caso de hiponatremia crónica, el manejo agresivo de reposición puede ser inapropiado ya que puede originar, incluso, la muerte. Las imágenes de radiografías de tórax con derrame pleural indican un mal manejo hidroelectrolítico, pues los datos de derrame desaparecen rápidamente y no parece tener corresponder a una infección nosocomial. Los diagnósticos finales se enlistan a continuación:
• Adolescente masculino con historia previa de intento de suicidio
• Transposición de colon
• Meningitis crónica, probablemente por tuberculosis
• Síndrome de secreción inapropiada de hormona antidiurética
Causa de la muerte: enclavamiento de amígdalas versus mielinolisis pontina o mesencefálica.
Terapia Intensiva (Dr. Adrián Chávez)
Es difícil que con solo utilizar la clínica se pueda apoyar un síndrome de secreción inapropiada de hormona antidiurética (SIHAD) o un síndrome cerebral perdedor de sal. Para algunos nefrólogos no existe tal diferencia de síndromes y los dos son uno mismo en diferentes momentos.
Es necesario señalar que el paciente con una lesión encefálica aguda de cualquier etiología más hiponatremia, al llegar a una sala de urgencias tiene un riesgo de muerte de 60% mayor que los que no tienen hiponatremia. Y no por la hiponatremia si no por la gravedad de la lesión que tiene ya que la hiponatremia es una parte más de la manifestación de gravedad. Si es un niño, requiere de una corrección urgente de la hiponatremia pues es lo que los mata.
El SIHAD es un estado hipervolémico hiponatrémico en el que se está perdiendo sal, natriuresis elevada. El perdedor de sal es un hipovolémico hipotónico o normotónico pero cuya diferencia se establece con un balance de sodio netamente negativo, perdiendo más sodio que el que se le administra. Para establecer la diferencia entre estos dos cuadros no basta la clínica. Se requiere de conocer la fracción excretada de urea, el balance de sodio de 24 horas, medir cuánto sodio recibe y pierde para realizar un índice de excreción de sodio.
El manejo inicial de hiponatremia con lesión encefálica aguda requiere siempre de corrección de sodio, no importa la causa.
Nefrología (Dr. Ricardo Muñoz Arizpe)
Considero que el manejo hidroelectrolítico fue deficiente, pues ingresó con acidosis metabólica por retención de hidrogeniones. Pero no se corroboró porque no se hizo una gasometría, aunque el pH urinario estaba en 5 con cuerpos cetónicos elevados de 150 mg/dl que indicaban una cetoacidosis no diabética, pero sí por ayuno prolongado de 1 a 2 semanas.
Además, se dio tratamiento a una hipocalcemia que no se comprobó, pues no se determinó albúmina sérica, por lo que sugiere a todos los médicos siempre medir también la albumina sérica para interpretar adecuadamente los valores de calcio.
La hiponatremia probablemente tenía una evolución de 1 a 2 semanas y, en presencia de osmolaridad urinaria de 770 y 240 mOsm, se concluye que el paciente no estaba deshidratado, lo que hace un diagnóstico inconfundible de síndrome de secreción inadecuada de hormona antidiurética. Hay más datos, como urea de 10 mg que no es normal para un niño de 14 años, y se explica porque estaba diluido. Esto marca la diferencia entre el SIHAD y el síndrome perdedor de sal. También, al revisar la velocidad de filtración glomerular de 182ml/min/1.73, se puede ver que tenía el doble para su edad. Esto significa que estaba extrafiltrando por el exceso de líquido extracelular, lo que sucede solamente en SIHAD. El manejo de este tipo de síndrome no es con sodio. El problema real es la retención de agua y sal. Solo debe de darse sodio en caso de crisis convulsivas, utilizando sodio hipertónico al 3% para aumentar la hipotonicidad y en una fase posterior una restricción de sodio. Este paciente cursó con remisiones temporales de la SIHAD, para exacerbarse con una hiponatremia grave, la cual trató de corregirse administrando más sodio que lo llevó a un aumento en la excreción urinaria del mismo y más hiponatremia, hipertensión arterial y, seguramente, hemorragia cerebral.
Patología (Dra. María Argelia Escobar Sánchez)
Antes de mostrar los hallazgos histopatológicos, les voy a mostrar la biopsia transoperatoria (Figura 6). Se recibieron, en fresco para estudio transoperatorio, varios fragmentos de tejido que medían en conjunto 4 x 4 x 0.5 cm. Se identificaban algunos más claros, necróticos, otros con áreas congestivas. Lo que se observó en los cortes definitivos teñidos con hematoxilina y eosina, fueron fragmentos de corteza cerebral y sustancia blanca. Se observó el espacio subaracnoideo con numerosos vasos sanguíneos, principalmente arteriolas de pequeño calibre con necrosis de su pared y extenso infiltrado inflamatorio constituido por linfocitos e histiocitos epitelioides (Figura 7). Había también algunas áreas con necrosis. Estas mismas características que se observaron en los vasos subaracnoideos estaban también en los vasos parenquimatosos. Se pudo observar una arteria de mediano calibre y fue evidente el infiltrado inflamatorio localizado en toda su pared. Se observó también el desprendimiento del endotelio y, en las zonas cercanas a esta arteria, necrosis y zonas de infarto (Figura 8).



Había también zonas de necrosis e infarto adyacente a los vasos sanguíneos. Nuevamente, la lesión vascular que se observó es muy evidente con presencia de necrosis fibrinoide en la luz. Se realizaron múltiples tinciones para la búsqueda de microorganismos, específicamente hongos, las cuales fueron negativas. Se realizó tinción de Ziehl Neelsen para búsqueda de bacilos ácido-alcohol resistentes (BAAR) (Figura 9). Se realizó el diagnóstico de meningoencefalitis granulomatosa.

Posteriormente, falleció el paciente. El habitus exterior muestra déficit de 8 kg, ya que tenía el antecedente de la ingestión de sosa caústica con la transposición colónica y la gastrostomía permanente, una traqueostomía y una cicatriz antigua de 26 cm, además de extremidades hipotróficas. Se observaron también heridas quirúrgicas recientes suturadas donde se realizó la ventriculostomía y la toma de biopsia.
El encéfalo pesó 1,500 g contra un esperado de 1,450 g. Llamó la atención, visto por la convexidad, el aspecto de las meninges. Estas se observaron opacas, despulidas, y se observó el orificio de la ventriculostomía. Visto por la base, las meninges se encontraron muy engrosadas, es decir, afectada toda la porción infratentorial. Presentó una meningitis fibrinopurulenta que es evidente desde el punto de vista macroscópico (Figura 10). Se realizaron cortes axiales, donde se evidenció que este paciente tenía un daño cerebral muy extenso de predominio en el hemisferio derecho, afectando frontal, temporal y ganglios de la base. Había extensas áreas de infarto y de necrosis que afectaron todo el mesencéfalo, puente y parte de los hemisferios cerebrales (Figura 11).


La lesión histológica se caracterizó por una vasculitis granulomatosa necrosante que afecta arteriolas y arterias de pequeño y mediano calibre. Estaba constituida por granulomas con numerosos linfocitos e histiocitos epitelioides que afectan toda la pared de los vasos sanguíneos. Además, hay pérdida por necrosis. Este infiltrado se extiende a las áreas perivasculares. En las zonas cercanas hay extensas áreas de hemorragia, necrosis, infartos hemorrágicos e isquémicos (Figura 12). En los vasos más afectados es evidente la vasculitis que presentaba este paciente. Se observa una vasculitis de larga evolución con lesiones en varias etapas de evolución. Se pueden ver lesiones plexiformes, nuevamente el infiltrado y la necrosis. A mayor aumento, la presencia de necrosis y numerosos histiocitos epitelioides que conforman los granulomas. Esta es una lesión histológica muy representativa de una vasculitis necrosante granulomatosa, donde podemos ver pérdida de la elástica interna y de la elástica externa por necrosis fibrinoide, además del infiltrado inflamatorio. En una tinción de PAS, que tiñe membranas basales, se puede observar la presencia de lesiones plexiformes y necrosis fibrinoide. Había otras arterias y arteriolas mucho más afectadas (Figura 13). Se realizaron tinciones para la búsqueda de microorganismos, especialmente hongos y micobacterias. Existen cinco causas de granulomas: micobacterias, hongos, enfermedades reumatológicas, reacciones a cuerpo extraño y sarcoidosis. De este grupo, las únicas que pueden dar granulomas con vasculitis son las causas reumatológicas. Estas son arterias y arteriolas de mayor calibre que podemos ver no están afectadas, el infiltrado inflamatorio es perivascular, la pared está íntegra.


Ahora se muestran los hallazgos en los pulmones. Estos tenían un peso un poco aumentado, de 750 g contra un esperado de 715 g. Macroscópicamente mostraron algunas áreas de congestión y dilatación de la trama bronquial. Histológicamente, se observa la misma lesión que en el sistema nervioso central, representada por vasculitis con granulomas, con infiltrado inflamatorio constituido por linfocitos e histiocitos epitelioides (Figura 14). Son evidentes los granulomas y se observa necrosis de la pared. Estas son lesiones que ocluyen la luz y dañan la pared de los vasos. Hay presencia de células gigantes multinucleadas con necrosis y fibrosis. No se observaron granulomas que sugieran tuberculosis. En una tinción de Masson se puede evidenciar que hay fibrosis extensa que habla de daño crónico con fibrosis intersticial y nuevamente las lesiones plexiformes en todas las arterias y arteriolas tanto en el pulmón derecho como en el pulmón izquierdo. En otras áreas hay, además, extenso infiltrado inflamatorio constituido por leucocitos polimofornucleares dentro de la luz de los alveolos, en los tabiques interalveolares y alrededor de los bronquiolos. Este paciente presenta una bronconeumonía con extensas áreas de hemorragia, infiltrado inflamatorio además de necrosis, detritus celulares y formación incipiente de membranas hialinas (Figura 15). Además de la bronconeumonía hay depósito de tejido conectivo alrededor de los bronquiolos, es decir el paciente tiene una bronquiolitis obliterante.


Se considera que los diagnósticos finales son los que a continuación se mencionan. Enfermedad principal: vasculitis necrosante granulomatosa de pequeños y medianos vasos, compatible con granulomatosis de Wegener que afecta el sistema nervioso central y ambos pulmones.1-5
Con base en el estudio de la autopsia y teniendo como sospecha una enfermedad reumatológica, además de haber descartado todas las causas infecciosas, se buscó intencionadamente y se realizó inmunofluorescencia, tanto en los riñones como en el sistema nervioso central y en los pulmones, donde se observó depósito de fibrinógeno l. En la propuesta de granulomatosis de Wegener no hay depósito de complejos inmunes. Se puede observar un depósito variable de IgG e IgM en pequeñas cantidades. Con todos estos hallazgos del estudio postmortem se buscó otra forma de estudiar el caso y se encontró un artículo en el que se demuestra que estos pacientes pueden tener un aumento de linfocitos T CD25 y CD4 positivos, y se propone que hay una alteración en la regulación de estos linfocitos.6 Nosotros realizamos CD25, CD4, CD8 y CD3, siendo CD8 y CD3 negativos, y observándose positividad para CD25 y CD4 y, efectivamente, se observó la sobreexpresión de linfocitos CD25, CD4 con disminución de linfocitos CD3 y CD8.
Con base en lo anterior, las alteraciones concomitantes fueron:
• Bronquiolitis obliterante
• Status post ventriculostomía derecha
• Status post toma de biopsia cerebral
• Status post traqueostomía
• Derrame pleural bilateral (500 ml D, 450 ml I)
• Desnutrición grado III (38.2 kg vs 50 kg)
En el resto de los órganos se observó lo siguiente. En el corazón se puede ver hipertrofia de los cardiomiocitos, por lo que el paciente seguramente tenía una insuficiencia cardiaca. Las coronarias no mostraron vasculitis.
El hígado tuvo un peso de 1,500 g contra un esperado de 1,200 g. Tenía una hepatomegalia congestiva. Histológicamente no había alteraciones en los espacios porta ni en el lobulillo, únicamente datos de congestión y extravasación de eritrocitos. Había, además, datos de choque como edema intraalveolar en los pulmones, presencia de necrosis tubular aguda en los riñones y trombos de fibrina en la microvasculatura de varios órganos. Con estos hallazgos se integraron los siguientes diagnósticos:
• Hepatomegalia congestiva (1,500 g vs 1,200 g)
• Datos anatómicos de choque:
- Necrosis tubular aguda
- Edema intraalveolar
- Trombos de fibrina en la microvasculatura de los pulmones y piel
Este paciente, como alteración independiente que no contribuye a la enfermedad principal, tenía el antecedente de una transposición colónica. Podemos ver que había engrosamiento de la serosa cuando se hizo la apertura del colon, vimos que estaba engrosada la pared y disminuida la luz. Histológicamente el colon mostró congestión en la lámina propia y a nivel de la submucosa.
Esta es la pieza que se disecó posteriormente, se dejó el esófago y el estómago, se ve la presencia de la gastrostomía. Estos son los cortes seriados donde se observaba el esófago con una luz de 0.1 cm de diámetro, tenía una estenosis de 100% y una pared totalmente fibrótica (Figura 16).

En los cortes mejor conservados, vemos el epitelio intacto y fibrosis de la pared. Los cortes de la gastrostomía con extenso infiltrado inflamatorio constituido por linfocitos, neutrófilos, había también células gigantes multinucleadas tipo reacción a cuerpo extraño y áreas de necrosis, constituyendo una peritonitis crónica secundaria a todo el proceso inflamatorio. Los cortes del estómago mostraron epitelio íntegro sin alteraciones, los cortes del intestino delgado únicamente mostraron áreas de congestión en la submucosa. Las alteraciones independientes fueron estas:
• Estenosis esofágica de 100% secundaria a ingestión de sosa caústica (en el año 2009)
• Status postransposición colónica (enero 2011)
• Statuts postgastrostomía (enero 2011)
• Peritonitis crónica con reacción granulomatosa a cuerpo extraño
Se estudiaron, intencionadamente y especialmente, los riñones en búsqueda de vasculitis. Estos tuvieron un peso dentro de lo esperado. La mucosa de la vejiga mostró congestión. Macroscópicamente, bien delimitada la corteza de la médula renal. Los riñones mostraron congestión a nivel de los glomérulos, en el intersticio y en los túbulos. No hay datos de vasculitis ni depósito de complejos inmunes. Revisando la literatura, está descrito que en adolescentes en ocasiones es rara la presentación renal en la granulomatosis de Wegener y está descrito que con alteraciones en el sistema nervioso central y en los pulmones se puede hacer diagnóstico.7
El bazo con un peso de 170 g contra un esperado de 110 g. Histológicamente de características normales, únicamente con congestión de la pulpa roja.
La médula ósea con celularidad normal. Esto se menciona porque muchos de los pacientes con enfermedad reumatológica presentan alteraciones de la médula ósea. El sistema endocrino tampoco presentó alteraciones. En los cortes del páncreas se observa con características normales, la glándula suprarrenal, la glándula tiroides y las gónadas sin alteraciones.
Estos son los cultivos post mortem:
• Hemocultivo, LCR, pulmón derecho: negativos
• Pulmón izquierdo, bazo, Intestino delgado: Escherichia coli
• Colon: Escherichia coli (dos morfotipos) y levaduras.
Al hacer una revisión en el hospital se encontró que, de 832 autopsias, nunca se había diagnosticado postmortem una granulomatosis de Wegener.
Reumatología (Dra. Rocío Maldonado Velázquez)
La enfermedad de Wegener es muy rara. Solo hay 7 casos manejados en el servicio, y este es el primer caso con daño en sistema nervioso central sin manifestación en otros sitios. La presentación más común es con daño renal y pulmonar. El 95% de pacientes con Wegener debutan con alteración respiratoria y el 70% con una lesión renal o la desarrolla en los primeros 2 años. Solo se reportan tres casos en niños con una presentación en sistema nervioso central, de los cuales todos fueron descubiertos en autopsias. Wegener es difícil de sospechar al no existir datos respiratorios.
Coordinador (Dr. Rómulo Erick Rosales Uribe)
Esta sesión nos ha dejado una gran enseñanza. Sin embargo, en nuestro medio, la presentación de enfermedad de Wegener es extremadamente rara, por lo que ante un caso de meningitis crónica con dilatación ventricular, pleocitosis en LCR a expensas de mononucleares, hiperproteinorraquia e hipoglucorraquia, se deberá, en primer lugar, sospechar de una tuberculosis meníngea, y el diagnóstico de enfermedad de Wegener será por exclusión.
REFERENCIAS
1. Polychronopoulos VS, Prakash UB, Golbin JM, Edell ES, Specks U. Airway involvement in Wegener's granulomatosis. Rheum Dis Clin North Am 2007;33:755-775. [ Links ]
2. Finkielman JD, Lee AS, Hummel AM, Viss MA, Jacob GL, Homburger HAS, et al. ANCA are detectable in nearly all patients with active severe Wegener's granulomatosis. Am J Med 2007;120: 643.e9-e14. [ Links ]
3. Flores-Suárez LF, Villa AR. Spectrum of Wegener granulomatosis in a Mexican population. Ann NY Acad Sci 2007;1107:400-409. [ Links ]
4. Moore BM, Rothman SM, Clark HB, Vehe RK, Laguna TA. Epilepsy: an anticipatory presentation of pediatric Wegener's granulomatosis. Pediatr Neurol 2010;43:49-52. [ Links ]
5. Dörr J, Elitok S, Dieste FJ, Vogel HP, Schneider W, Wuerfel J, et al. Treatment-resistant chronic headaches and focal pachymeningitis in a 46-year-old man: a rare presentation of Wegener's granulomatosis. Lancet Neurol 2008;7:368-372. [ Links ]
6. Morgan MD, Day CJ, Pipper KP, Khan N, Harper L, Moss PA, et al. Patients with Wegener's granulomatosis demonstrate a relative deficiency and functional impairment of T-regulatory cells. Immunology 2010;130:64-73. [ Links ]
7. D'Hauwe R, Lerut E, Breysem L, Smet M. A rare presentation of renal Wegener's granulomatosis in a child. Pediatr Radiol 2011;41:1212-1215. [ Links ]

