










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.70 no.4 México jul./ago. 2013
CASO CLÍNICO
Oclusión de colateral aorto-pulmonar con dispositivo Amplatzer® Vascular Plug II en un paciente con atresia pulmonar y comunicación interventricular. Reporte de un caso
Occlusion of aortopulmonary collateral arteries with Amplatzer® Vascular Plug II device in a patient with pulmonary atresia and ventricular septal defect. Case report
Luis Alexis Arévalo-Salas,1 Ada Lila Lacayo-Molina,1 Jorge Luis Villatoro-Fernández,1 Rosa Aurora Lozano-Díaz,1 Alejandro Bolio-Cerdán,2 Julio Erdmenger-Orellana1
1 Servicio de Hemodinámica, Departamento de Cardiología
2 Departamento de Cirugía Cardiovascular, Tórax y Endoscopia
Hospital Infantil de México Federico Gómez. México D.F., México
Autor de correspondencia:
Dr. Luis Alexis Arévalo Salas
Correo electrónico: alexisarevalosalas07@gmail.com
Fecha de recepción: 20-05-13
Fecha de aceptación: 04-07-13
Resumen
Introducción. La atresia pulmonar con comunicación interventricular es una cardiopatía compleja con una incidencia aproximada de 2% entre todas las cardiopatías congénitas. Se asocia con frecuencia al síndrome de deleción 22q11. Tiene una amplia variabilidad anatómica que es necesario precisar con exactitud para poder establecer un plan médico quirúrgico individualizado.
Caso clínico. Se presenta el caso de un paciente de 2 años con atresia pulmonar y comunicación interventricular asociadas a dos grandes colaterales aortopulmonares. Este paciente fue corregido mediante conexión de ventrículo derecho a arteria pulmonar, durante la cual fue posible ligar una de las colaterales; la restante fue embolizada mediante un dispositivo trascateterismo con oclusión total.
Conclusiones. El manejo de las arterias colaterales asociadas a atresia pulmonar con comunicación interventricular es complejo pero accesible al tratamiento trascateterismo mediante el implante de dispositivos con mínima morbimortalidad.
Palabras clave: embolización de colaterales trascateterismo, atresia pulmonar, colaterales aortopulmonares.
Abstract
Background. Pulmonary atresia with ventricular septal defect (VSD) is a complex heart disease with an incidence of ~2% of all congenital heart diseases. It is frequently associated with 22q11 deletion syndrome. Due to the extensive anatomic variability, it is necessary to accurately establish an individualized surgical/medical plan.
Case report. We report the case of a 2-year-old patient with pulmonary atresia and two associated mayor aortopulmonary collateral arteries. This patient underwent right ventricular-pulmonary artery connection at which time it was possible to ligate one of the collaterals. The remaining were embolized by transcatheter device with total occlusion.
Conclusions. Management of collateral arteries associated with pulmonary atresia with VSD is complex but is accessible with transcatheter treatment with device implantation with minimal morbidity.
Key words: transcatheter embolization of collaterals, pulmonary atresia, aortopulmonary collaterals
Introducción
La atresia pulmonar con comunicación interventricular (AP+CIV) puede considerarse como la variedad más grave de la tetralogía de Fallot aunque la anatomía suele ser más compleja y, por ende, es necesario un estudio anatómico preciso que permita establecer un programa terapéutico médico-quirúrgico adecuado para cada paciente. La AP+CIV tiene una incidencia de 2% entre todas las cardiopatías congénitas.1 La variabilidad anatómica incluye hipoplasia de tronco y ramas pulmonares, anormalidades de la vasculatura pulmonar y la presencia de circulación anormal proveniente, en la mayoría de las veces, de la aorta descendente por grandes vasos, generalmente entre 1 y 8.2,3 Estos brindan perfusión a diversas porciones de ambos pulmones. En ocasiones pueden contribuir a incrementar el flujo pulmonar. El acceso quirúrgico para la ligadura o sección de estas colaterales puede ser complicado, por lo que se pueden implementar procedimientos trascateterismo para la oclusión de esas arterias anormales, particularmente si son clínicamente significativas.
La meta de la cirugía debe ser conectar el ventrículo derecho con la arteria pulmonar y, en caso de existir arterias colaterales, conectar a la circulación pulmonar central el mayor número posible de estas (focalización) o bien, si la conexión ventrículo derecho-arteria pulmonar es suficiente, descartar a la circulación colateral mediante cirugía o embolismo. Desde el año 2000 la Congenital Heart Surgeons Society publicó la caracterización de la circulación pulmonar con base en los hallazgos angiográficos, donde se establece una guía terapéutica de acuerdo con la anatomía y fisiología individualizada.4
Cabe destacar que la AP+CIV se asocia fuertemente con el síndrome de deleción 22q11, particularmente cuando están presentes grandes colaterales.5 Por ello, resulta obligada la búsqueda de la facies característica, así como el estudio cromosómico, puesto que la presencia de este síndrome tiene grandes implicaciones en el futuro de los niños.
Presentamos el caso de un paciente con AP+CIV que fue operado mediante conexión ventrículo pulmonar con injerto protésico. Posteriormente se practicó la oclusión de una gran arteria colateral de aorta descendente a la circulación pulmonar mediante un dispositivo Amplatzer® Vascular Plug II.
Caso clínico
Se trató de un preescolar masculino de 36 meses de edad, producto del tercer embarazo normoevolutivo. Se obtuvo por parto eutócico, peso de 2,690 g, talla de 52 cm y Apgar de 8-9. Sin antecedentes heredofamiliares de interés para su padecimiento. Se detectó cianosis peribucal y ungueal que se incrementaba al llanto desde el primer mes de vida y dificultad respiratoria asociada a tiraje intercostal. El paciente fue referido a nuestro instituto a los 10 meses de edad. A su ingreso se detectó que no había una facies anormal, cianosis grado II, hiperactividad precordial izquierda y soplos continuos en la periferia de ambos campos pulmonares con intensidad II/VI. El segundo ruido era único. Se practicaron estudios de gabinete que descartaron la deleción 22q11 y en los que se reportó atresia pulmonar con comunicación interventricular, con un cabalgamiento de 50%, masas infundibulares obstruyendo el tracto de salida ventricular derecho y atresia valvular con hipoplasia grave de anillo pulmonar. Las ramas pulmonares eran confluentes: la derecha, de 9.68 mm de diámetro (Z=+2) y la izquierda, de 9.49 mm (Z=+1). Un primer cateterismo cardiaco practicado al año de edad mostró dos grandes colaterales. Una de ellas nacía de la subclavia derecha y comunicaba hacia ambas ramas de arteria pulmonar (Figura 1). La otra colateral nacía de la aorta descendente (con diámetro de 5 mm) y comunicaba flujo directamente a la rama derecha de la arteria pulmonar. La presión en estos dos sistemas era similar en 44/26 mmHg (con media de 34 mmHg). Con estos hallazgos se decidió practicar una corrección total a los 2 años de edad. Durante la cirugía se confirmaron los datos señalados en el cateterismo cardiaco. Se colocó un parche para el cierre de la comunicación interventricular, ligadura de la colateral dependiente de subclavia derecha y se implantó un tubo valvulado Hancock® de 16 mm de diámetro entre el infundíbulo ventricular derecho y la arteria pulmonar, sin complicaciones. La evolución ulterior fue satisfactoria. Sin embargo, persistió con discreta polipnea en los meses siguientes a la cirugía. Nueve meses después en consulta de revisión, se confirmó la polipnea. Un ecocardiograma de control reveló un flujo importante por la colateral de aorta descendente, por lo que se programó un cateterismo cardiaco para diagnóstico y para valorar la posibilidad de oclusión de esta.

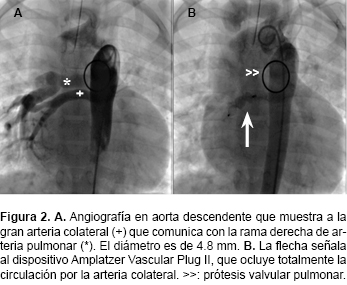
El segundo cateterismo mostró un salto de saturación de 15% (66% en venas cavas a 81% en rama derecha de arteria pulmonar) con presión de 30/10 mmHg (media de 12 mmHg) en rama derecha de arteria pulmonar. La relación de gastos era de 1.8:1 ocasionado por la arteria colateral comunicante; el diámetro máximo de esta era de 4.8 mm por lo que se decidió su oclusión con un dispositivo Amplatzer® Vascular Plug II de 8 mm (Figura 2). Inmediatamente después de la oclusión, la saturación en la rama derecha se normalizó. La presión en esta rama disminuyó a 17/7 mmHg (media de 10 mmHg) y la relación de gastos se estabilizó en 1:1. No hubo complicaciones durante el procedimiento y el paciente fue egresado el mismo día.
Discusión
Las arterias colaterales aortopulmonares de gran tamaño que comunican la circulación sistémica con la pulmonar son un hallazgo frecuente en AP+CIV y representan el remanente de arterias esplácnicas ventrales embrionarias, que en condiciones normales deben mostrar una regresión conforme se establece la circulación arterial pulmonar en las primeras semanas de gestación.6 Sin embargo, estas arterias se convierten en arterias colaterales de diverso tamaño y funcionalidad con relación directa a un mal desarrollo de la circulación arterial pulmonar, específicamente en casos de atresia pulmonar. Y esto deriva en una amplia gama de alteraciones circulatorias pulmonares que van desde la simple atresia pulmonar con ramas confluentes, hipoplasia de ramas pulmonares, pérdida de la confluencia de las arterias pulmonares, presencia de grandes colaterales aortopulmonares que pueden o no ser comunicantes con la circulación pulmonar hasta la agenesia de ramas pulmonares verdaderas. Es evidente que cada una de las variedades anatómicas posibles necesita de un diseño individualizado para la corrección quirúrgica, cuya meta principal es restablecer, en la medida de lo posible, la circulación ventrículo-pulmonar. Pero esto dependerá de las características anatómicas de las ramas pulmonares y de las arterias colaterales. Las opciones, por tanto, son restablecer la continuidad ventrículo pulmonar mediante un parche o un tubo valvulado (de acuerdo con la anatomía intracardiaca), la focalización total o por estadios (de acuerdo con la anatomía de ramas pulmonares y de las arterias colaterales) o decidir el cierre o no de la comunicación interventricular (según la hipoplasia de las ramas pulmonares).7-9
En este caso, la anatomía ventricular y de las ramas pulmonares permitió llevar a cabo una conexión del ventrículo derecho a la arteria pulmonar en la que se implantó un tubo valvulado. Durante el procedimiento quirúrgico fue asequible ligar la gran colateral dependiente de la subclavia derecha. Sin embargo, fue difícil eliminar quirúrgicamente la colateral derivada de la aorta descendente. Durante el seguimiento fue evidente que generaba hiperaflujo pulmonar, ya que se detectó un QP/QS de 1.8:1 y un salto de saturación de 15% entre la vena cava superior y la arteria pulmonar derecha, por lo que se decidió el cierre trascateterismo de la colateral. Dado el diámetro de 4.8 mm se consideró que la mejor opción era utilizar un dispositivo Amplatzer® Vascular Plug II de 8 mm de diámetro, que permitió el cierre total, inmediato y sin complicaciones de esta colateral, normalizando la circulación pulmonar.
Los pacientes con AP+CIV pueden presentar múltiples y grandes arterias colaterales sin obstrucción, como en este caso, lo que produce un excesivo aflujo de sangre a los pulmones y predispone a insuficiencia cardiaca. Por esto, resulta necesario intervenirles para suprimir esa circulación. La forma tradicional es la ligadura de las colaterales. Sin embargo, en ocasiones, pueden originarse de sitios difíciles o alejados del campo operatorio y es cuando la alternativa trascateterismo es útil. Históricamente, se han utilizado diversos materiales para embolizar, incluyendo adhesivos tisulares como el Gelfoam® (gelatina de piel porcina purificada), balones de silicón y espirales de Gianturco.10 En la actualidad es posible que estos últimos sean los de uso más extendido. Se ha reportado como un procedimiento exitoso en más de 90% de los casos.6 Sin embargo, en algunas ocasiones, por la posición de los vasos, el diámetro y su propia tortuosidad, el implante puede ser difícil o requerir de varias espirales.
El desarrollo de nuevos instrumentos, como los dispositivos Amplatzer® Vascular Plug (AVP) elaborados con una malla de níquel y titanio (nitinol) con memoria auto expandible,11 ha permitido simplificar la oclusión de casos seleccionados de diversas malformaciones arterio-venosas, incluyendo a las grandes colaterales relacionadas a la AP+CIV. En la actualidad, la versión moderna del AVP es la II que fue aprobada por la FDA en 2007 para su empleo clínico.12 Esta nueva versión consta de tres segmentos con seis capas densas de nitinol, que brinda mejores propiedades oclusivas. Además, el implante del dispositivo es sencillo y mediante catéteres de perfil bajo. Por otro lado, la complicación más temida, que es la migración del aparato, se minimiza ya que al ser autoexpandible genera la suficiente fuerza radial para mantenerse inmóvil una vez desplegado.
En los casos de AP+CIV con grandes colaterales aortopulmonares existe una estrecha relación con el síndrome de deleción 22q11. Existe cierta evidencia clínica que relaciona a las colaterales que nacen de los vasos braquiocefálicos con este síndrome en mayor proporción que aquellos con grandes colaterales que nacen directamente de la aorta descendente.13 Esta información es importante, particularmente por la historia natural de los pacientes con deleción 22q11, no solamente en el aspecto cardiológico sino en el neurológico y psiquiátrico. En este caso, esta posibilidad fue descartada por el estudio de FISH.
Es importante referir que la evolución a largo plazo de las colaterales es incierta y pueden condicionar enfermedad vascular pulmonar en algunos casos, o bien, evolucionar a estenosis progresiva con daño pulmonar paulatino. Por esto mismo, restablecer la circulación pulmonar de la manera más fisiológica posible es mandatorio en la AP+CIV.
REFERENCIAS
1. O'Leary P, Mair D, Edwards W, Julsrud P, Puga F. Pulmonary atresia and ventricular septal defect. En: Allen H, Gutgesell H, Clark E, Driscoll D, eds. Moss and Adams' Heart Disease in Infants, Children and Adolescents. Including the Fetus and Young Adult. Philadelphia: Lippincott, Williams and Wilkins; 2001. pp. 864-879. [ Links ]
2. McGoon MD, Fulton RE, Davis GD, Ritter DG, Neill CA, White RI Jr. Systemic collateral and pulmonary artery stenosis in patients with congenital pulmonary valve atresia and ventricular septal defect. Circulation 1977;56:473-479. [ Links ]
3. Hoffman J: Tetralogy of Fallot with pulmonary atresia. En: Hoffman J, ed. The Natural and Unnatural History of Congenital Heart Disease. West Sussex: Wiley-Blackwell; 2009. pp. 437-445. [ Links ]
4. Tchervenkov CI, Roy N. Congenital Heart Surgery Nomenclature and Database Project: pulmonary atresia-ventricular septal defect. Ann Thorac Surg 2000;69(suppl 4):S97-S105. [ Links ]
5. Frohn-Mulder IM, Wesby Swaay E, Bouwhuis C, Van Hemel JO, Gerritsma E, Niermeyer MF, et al. Chromosome 22q11 deletions in patients with selected outflow tract malformations. Genet Couns 1999;10:35-41. [ Links ]
6. Boshoff D, Gewillig M. A review of the options for treatment of major aortopulmonary collateral arteries in the setting of tetralogy of Fallot with pulmonary atresia. Cardiol Young 2006;16:212-220. [ Links ]
7. Seok Bang J, Suk Baek J, Zhu L, Bae E, Il Noh Ch, Choi JY, et al. Pulmonary atresia with ventricular septal defect and major aorto-pulmonary collateral arteries: management strategy at our hospital and the results. Korean Circulation J 2007;37:348-352. [ Links ]
8. Balaguru D, Dilawar M. Pulmonary atresia with ventricular septal defect: systematic review. Heart Views 2005;8:52-61. [ Links ]
9. Gupta A, Odim J, Levi D, Chang RK, Laks H. Staged repair of pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries: experience with 104 patients. J Thorac Cardiovasc Surg 2003;126:1746-1752. [ Links ]
10. Rao P. Transcatheter embolization of unwanted blood vessels in children. En: Rao P, Kern M, eds. Catheter Based Devices for the Treatment of Non-coronary Cardiovascular Disease in Adults and Children. Philadelphia: Lippincot Wiliams & Wilkins; 2003. pp. 457-473. [ Links ]
11. Küçükay F, Sarper R, Cumhur T. Three huge pulmonary arteriovenous malformations: endovascular embolization with the Amplatzer vascular plug. Tur Toraks Der 2011;12:168-171. [ Links ]
12. Wang W, Li H, Tam MD, Zhou D, Wang DX, Spain J. The Amplatzer vascular plug: a review of the device and its clinical applications. Cardiovasc Intervent Radiol 2012;35:725-740. [ Links ]
13. Sureka A, Peng L, Reinhartz O, Reddy M, Hanley M. Anatomy in patients with 22q11 deletion and pulmonary atresia with ventricular septal defect and major aortopulmonary collaterals. Surg Sci 2011;2:294-296. [ Links ]

