










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versão impressa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.69 no.2 México Mar./Abr. 2012
Tema pediátrico
Manifestaciones dermatológicas del síndrome de Alagille
Dermatologic manifestations of Alagille syndrome
André Morales Martínez,1 Carlos Alfredo Mena Cedillos,1 Jaime Nieto Zermeño,2 Verónica Morán Barroso,3 Salvador Villalpando Carrión,2 Silvia Ramírez Dovala4
1 Servicio de Dermatología Pediátrica, Hospital Infantil de México Federico Gómez
2 Dirección de Enseñanza y Asistencia Médica, Hospital Infantil de México Federico Gómez
3 Departamento de Genética, Hospital Infantil de México Federico Gómez
4 Departamento de Dermatología, Hospital General de México, México D.F., México
Autor de correspondencia:
Dr. André Morales Martínez
Correo electrónico: doctor_andre@yahoo.com
Fecha de recepción: 12-01-11.
Fecha de aceptación: 31-01-12.
Resumen
El síndrome de Alagille (ALGS1; MIM118450) es la causa de una gran parte de las colestasis de origen congénito. Se trata de una enfermedad multisistémica con un patrón de herencia autosómico dominante y manifestaciones clínicas variables. Está conformado por los criterios clásicos de colestasis, defectos cardiacos, anormalidades óseas, oculares y rasgos faciales característicos. Se ha asociado a mutaciones del gen JAG1, con locus en 20p12.2.
Existe una segunda forma del síndrome de Alagille (ALGS2; MIM610205) causada por mutaciones en el gen NOTCH2, con locus en 1p13-p11. El diagnóstico puede ser difícil de establecer ya que no todos los pacientes muestran un cuadro clásico en forma temprana, por lo que aumenta la morbilidad del padecimiento. En esta revisión se proponen herramientas clínicas para sospechar de este síndrome y abordarlo de manera precoz.
Palabras clave: síndrome de Alagille, colestasis, manifestaciones cutáneas.
Abstract
Alagille syndrome (MIM #118450) causes the majority of cases of congenital cholestasis. It is an autosomal dominant multisystem disorder associated with several different clinical manifestations including the core criteria of cholestasis, cardiac defects, skeletal abnormalities, and eye and facial features. This condition is caused by mutations in the JAG 1 gene on chromosome 20p12. It is known a second form of Alagille syndrome that is caused by mutations in the NOTCH gene on 1p13-p11 (MIM #610205). Diagnosis is often delayed because only few patients show a classical picture in the early stages of the disease, causing an increase in morbidity. This review suggests clinical tools for early suspicion and management.
Key words: Alagille syndrome, cholestasis, cutaneous manifestations.
INTRODUCCIÓN
El síndrome de Alagille (SA) o displasia arteriohepática congénita es una forma sindromática de colestasis crónica por hipoplasia de vías biliares intrahepáticas, que se asocia a malformaciones congénitas extrahepáticas en niños con un fenotipo peculiar. Presenta un patrón de herencia autosómico dominante de expresión variable, por alteraciones del gen JAG1 en el locus 20p12.2.1-4 Desde su descripción, en 1969, por Alagille y colaboradores, ha sido reportado como la causa más común de colestasis crónica en occidente.5 Clásicamente, el diagnóstico se basa en los hallazgos histopatológicos, que son la escasez de conductos biliares interlobulillares y tres a cinco de los criterios mayores: colestasis crónica, cardiopatía, anomalías esqueléticas, oculares y fenotipo característico.3 El SA es un padecimiento poco frecuente que se presenta en uno de cada 70,000 a 100,000 recién nacidos, sin preferencia de género. Algunos casos se han asociado a la deleción en el cromosoma 20p, otros a mutaciones de novo.6,7
Características clínicas del síndrome de Alagille
El SA expresa anormalidades hepáticas, esqueléticas, renales, oculares y faciales, por lo que existen criterios mayores y menores para establecer el diagnóstico. Estos se señalan a continuación.
Criterios mayores
• Colestasis crónica. Está asociada a pausicidad ductal (más de la mitad de espacios porta sin ductos biliares) o ausencia de ductos biliares intrahepáticos reportados en la biopsia hepática, y se acompaña de prurito e hipercolesterolemia
• Facies característica. Con hipertelorismo, frente amplia, mentón prominente, ojos hundidos, nariz bulbosa en silla o recta
• Anomalías vertebrales. Vértebras en mariposa, principalmente en el segmento torácico
• Embriotoxon ocular posterior. Se trata de una prominencia de la línea de Schwalbe que marca la terminación periférica de la membrana de Descement. Esta presente hasta en 90% de los casos. Con menor frecuencia se encuentra exotropia. Es también criterio diagnóstico la presencia de pupila ectópica, banda queratósica y alteraciones del cristalino. Se sabe que el embriotoxon posterior puede presentarse como un rasgo autosómico dominante aislado; sin embargo, asociado al síndrome colestásico, da la pauta para sospechar de SA
• Cardiopatía congénita. La alteración más común es la estenosis arterial pulmonar periférica y, con menor frecuencia, la tetralogía de Fallot (7-9%), la persistencia del conducto arterioso, la comunicación interventricular, la comunicación interauricular, la atresia pulmonar y la coartación aórtica. Además, se han incluido algunas anomalías vasculares sistémicas como la estenosis de la arteria renal o carotidea
Criterios menores
• Xantomas secundarios a colestasis (por niveles de colesterol mayores a 500 mg/dl). Están localizados primordialmente en zonas de extensión, dedos, palmas, cuello, región inguinal y hueco poplíteo
• Peso y talla bajos para la edad, infecciones pulmonares recurrentes, insuficiencia pancreática (la cual debe ser sospechada ante cuadros diarreicos), hipotiroidismo, hipogonadismo, pubertad retrasada, retraso del desarrollo y voz atiplada-ronca
• Anomalías vasculares como la enfermedad de Moyamoya, alteraciones de los radicales venosos portales intrahepáticos y anomalías vasculares cerebrales con riesgo de hemorragia intracraneal
• Alteraciones neurológicas como neuropatía periférica secundaria a déficit de vitamina E2,3
• Alteraciones renales como nefrolitiasis, defectos en la concentración de orina, hipoplasia, duplicidad, quistes, riñón único, pelvis bífida, riñón ectópico, insuficiencia renal y nefropatía túbulo intersticial1-7
• Anteriormente, se consideraba que estos pacientes presentaban retraso mental; sin embrago, ahora se sospecha que las deficiencias vitamínicas, en particular de vitamina E, sean la causa de esta situación clínica, ya que la mayoría de estos pacientes conservan una inteligencia normal3
• El diagnóstico definitivo se establece con base en la biopsia hepática, cuando se encuentra un mínimo de 20 espacios porta y una relación entre el número de conductos biliares y de espacios porta menor de 0.4 (valores normales de 0.9 a 1.8). También, se puede detectar fibrosis en grado variable3,5,8
• El análisis molecular permite detectar mutaciones en la secuencia del gen JAG1
No todos los pacientes con SA muestran todas las características ya mencionadas, aún las clásicas; hay algunos que inicialmente no presentan ictericia, ni ascitis ni xantomas. Esto dificulta y retrasa por años la sospecha diagnóstica, por lo que incrementa la morbimortalidad del padecimiento.3,5,8
Manifestaciones clínicas del síndrome de Alagille
El objetivo de esta revisión fue evaluar las manifestaciones clínicas del SA, que son prácticamente específicas de esta enfermedad y, en general, poco consideradas dentro del abordaje diagnóstico. Si bien se le resta importancia a lo que es un elemento de apoyo relevante ante la gran gama de formas clínicas de esta enfermedad, la sumatoria de estos elementos es la clave para dar al clínico la sospecha diagnóstica, y con ello el inicio temprano del estudio y manejo de esta entidad, para mejorar el pronóstico y calidad de vida del paciente.
Las manifestaciones cutáneas pueden ser consideradas como la expresión de las diferentes alteraciones internas del SA. Se presentan desde el nacimiento, como los pliegues interdigitales extranumerarios y la facies característica. Generalmente les siguen otras manifestaciones, como la ictericia, el prurito, la liquenificación y los xantomas. Después se presentan las alteraciones secundarias a la afección vascular y a la desnutrición, motivos por los que surge la inquietud de la familia por solicitar atención médica. Ante lo poco frecuente de esta patología, los síntomas podrían pasar por otra condición, diferente o transitoria (Cuadro 1).7,9,10

Las manifestaciones clínicas de todas las afecciones hepáticas son principalmente cutáneas.9-11 La obstrucción o pseudobstrucción prolongada de las vía biliares determina la acumulación de colesterol en el suero. Se trata de colesterol no esterificado en su mayor parte, que estimula la formación de una lipoproteína anormal o lipoproteína X. La hipercolesterolemia prolongada conduce a la formación de xantomas planos, xantelasmas y xantomas tuberosos, que pudieran confundirse con otras condiciones dermatológicas.12
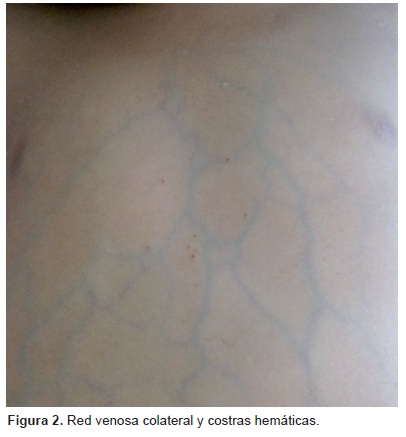
Además de la ictericia fuera del rango fisiológico (Figura 1), la acumulación de bilis condiciona un incremento de pruritógenos en el plasma. Son producidos en el hígado y excretados bajo condiciones fisiológicas, lo que conduce al incremento en los niveles opioides endógenos que finalmente inducen prurito. Este ha sido mencionado como el más severo de los síntomas en el SA. Incluso, debido a la molestia e incomodidad que genera para el paciente, llega a ser criterio para trasplante hepático en situaciones específicas.13 También se han mencionado como manifestaciones del SA, la xerosis, excoriaciones, telangiectasias, red venosa colateral (Figura 2), eritema palmar, alopecia, linfedema y queratodermias.

Con respecto a la dermatosis más representativa de esta entidad, García y colaboradores realizaron un estudio en el cual encontraron que 7 de 38 pacientes (29%) presentaron xantomas diseminados.10 Esto es similar a lo que se ha reportado en la literatura (entre 28 y 45%). Los xantomas se originan a partir de la colestasis crónica y la hipercolesterolemia, aunque no siempre se han relacionado los niveles elevados de colesterol a la aparición de dichas lesiones.8,13 Los xantomas también pueden observarse en los pabellones auriculares, la nuca, las superficies extensoras de los dedos, palmas, plantas, fosa poplítea, áreas inguinales, glúteos, codos y rodillas (Figura 3).

La edad más temprana de presentación del SA fue a los 10 meses, incrementando la severidad de acuerdo con el grado y la duración de la colestasis.3,4,8,11,12,14 Se han observado algunas peculiaridades de los xantomas: son confluentes en codos y rodillas, y debido al intenso prurito, se encuentra la presencia de liquenificación, excoriación y eczema. Su aparición advierte la necesidad de un trasplante hepático en más de 90% de los pacientes que los presentan debido a la progresión de la disfunción hacia la falla hepática.4,8,14 Tienden a desaparecer a la par de la disminución de los niveles séricos de colesterol, y con mayor rapidez después del trasplante hepático, después del cual se observa una mejoría dramática del prurito y de las lesiones cutáneas.3,10,14
En el estudio realizado por Kamath y colaboradores, se reporta la presencia de pliegues interdigitales supra-numerarios en 35% de la población de pacientes con diagnóstico de SA en el 2002. Esta presencia se asoció con una mutación del gen JAG1, aunque aún no es clara su función en el desarrollo normal de esta región (Figura 4).15

La identificación de las dermatosis presentadas en forma aislada es de gran importancia, tanto para la sospecha diagnóstica del SA como para otras causas de colestasis, de acuerdo con la edad y el resto del marco clínico en que se presente. Sin embargo, la asociación con los demás datos clínicos mencionados orienta fuertemente hacia el diagnóstico del síndrome.
La pronta identificación del SA ayudará a prevenir situaciones adversas que ponen en riesgo la vida del paciente, evitando así la morbilidad y, en gran medida, el uso de tratamientos que no resuelven ni mejoran la etiopatogenia específica.
El pronóstico de esta enfermedad está determinado por la alteración vascular asociada (Figura 5) y la degeneración producida por la colestasis, así como por los altos niveles de colesterol, además del riesgo de insuficiencia hepática y renal, que dan la pauta para establecer la gravedad del problema. En algunos de estos pacientes, la enfermedad degenera en hepatopatía terminal, por lo que requieren un trasplante hepático urgente. Otros pacientes mueren de forma temprana por infecciones o por afecciones cardiacas.10-16

El manejo de los pacientes con SA debe enfocarse en la corrección de las complicaciones de los sistemas afectados. Es de gran utilidad contar con el mayor número de herramientas diagnósticas que apoyen al médico tratante a inferir las diferentes complicaciones que el síndrome conlleva, para identificarlas desde etapas tempranas. De esta manera, se puede retrasar e, incluso, evitar la presentación de muchas de ellas, además de dar una mejor calidad de vida al paciente. También es fundamental proporcionar el adecuado consejo genético a los padres.2,8,15,16
REFERENCIAS
1. Online Mendelian Inheritance in Man. Alagille syndrome. Disponible en: http://www.omim.org/entry/118450. [ Links ]
2. Chen H. Alagille syndrome. En: Atlas of Genetic Diagnosis and Counseling. Totowa, NJ: Humana Press; 2006. pp. 32-35. [ Links ]
3. Rosenthal P. Atresia biliar y trastornos neonatales de las vías biliares. En: Wyllie R, Hyams J, eds. Gastroenterología Pediátrica. Philadelphia: McGraw-Hill Interamericana; 2001. pp. 640-641. [ Links ]
4. Ruiz-Castillo M, Michel-Penichet F, Cervantes-Bustamante R, Zarate-Mondragón F, Mata-Rivera N, Montijo-Barrios E, et al. Síndrome de Alagille: informe de 12 casos en el Instituto Nacional de Pediatría. Rev Enfer Infecc Pediatr 2007;81:13-17. [ Links ]
5. Wang JS, Wang XH, Zhu QR, Wang ZL, Hu XQ, Zheng S. Clinical and pathological characteristics of Alagille syndrome in Chinese children. World J Pediatr 2008;4:283-288. [ Links ]
6. Jiménez-Jiménez JR, Castellanos-Reyes K, Huerta-Albarrán R, Justiniani-Cedeño NE, Yáñez-López MP, Sierra-Tortolero A. Un caso del síndrome de Alagille. Rev Mex Pediatr 2007;74:152-157. [ Links ]
7. Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet 1997;34:152-157. [ Links ]
8. Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999;29:822-829. [ Links ]
9. Segupta S, Das JK, Gangopadhyay A. Alagille syndrome with prominent skin manifestations. Indian J Dermatol Venereol Leprol 2005;71:119-121. [ Links ]
10. García M, Ramonet M, Ciocca M, Cabrera H, Lapunzina P, Álvarez E, et al. Alagille syndrome: cutaneous manifestations in 38 children. Pediatr Dermatol 2005;22:11-14. [ Links ]
11. Ghosn SH, Kibbi AG. Cutaneous manifestations of liver diseases. Clin Dermatol 2008;26:274-282. [ Links ]
12. White LE. Xantomatosis y otras enfermedades de las lipoproteínas. En: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Lefell DJ, eds. Fitzpatrick Dermatología en Medicina General. Argentina: Médica Panamericana; 2009. pp 1272-1281. [ Links ]
13. Schwartz R, Rehder K, Parsons DJ, Morrell DS. Intense pruritus and failure to thrive in Alagille syndrome. J Am Acad Dermatol 2008;58(suppl 2):S9-S11. [ Links ]
14. Buckely DA, Higgins EM, du Vivier AW. Resolution of xanthomas in Alagille syndrome after liver transplantation. Pediatr Dermatol 1998;15:199-202. [ Links ]
15. Kamath BM, Loomes KM, Oakey RJ, Krantz ID. Supernumerary digital flexion creases: an additional clinical manifestation of Alagille syndrome. Am J Med Genet 2002;112:171-175. [ Links ]
16. Ling SC. Congenital cholestatic syndromes: what happens when children grow up? Can J Gastroenterol 2007;21:743-751. [ Links ]

