










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.67 no.6 México nov./dic. 2010
Caso clínico patológico
Adolescente con debilidad muscular proximal, eritema en heliotropo y neumonía intersticial aguda
Dermatomyositis, heliotrope erythema and acute interstitial pneumonia in an adolescent patient
Guillermo Ramón García,1 Enrique Faugier Fuentes2
1 Jefe de Servicio, Departamento de Patología
2 Jefe de Servicio, Departamento de Reumatología, Hospital Infantil de México Federico Gómez, México, D.F., México
Autor de correspondencia:
Dr. Guillermo Ramón García
Correo electrónico: gramon55@aol.com
Fecha de recepción: 20-07-10.
Fecha de aceptación: 20-07-10.
Resumen de la historia clínica
A-08-15
Paciente masculino, de 13 años 10 meses de edad, originario de Ciudad Sahagún, Hidalgo.
Motivo de consulta. Fiebre, tos, dificultad respiratoria.
Antecedentes heredo-familiares. Madre de 50 años, escolaridad preparatoria, ama de casa, hipertensión arterial sistémica desde los 20 años. Padre de 44 años, comerciante, con tabaquismo. Una hermana de 11 años en tratamiento por enfermedad reumatológica en otra institución. Diabetes mellitus tipo 2 e hipertensión arterial sistémica por rama materna. Con antecedente de cardiopatía no especificada y padecimientos dermatológicos no especificados por rama paterna.
Antecedentes no patológicos. Originarios del estado de Hidalgo, nivel socioeconómico medio. Buena alimentación en calidad y cantidad, inmunizaciones competas para la edad. Desarrollo psicomotor normal.
Antecedentes perinatales y patológicos. Producto GII, nació a término mediante cesárea por preeclampsia, sin complicaciones. Período neonatal sin complicaciones. Hernioplastia umbilical al año de edad. Hospitalizado a los 12 años por un mes en hospital de segundo nivel por neumonía, tratado con ampicilina y posteriormente ceftriaxona. Referido en Enero del 2008 a los 12 años 6 meses de edad al Hospital Infantil de México Federico Gómez por edema de manos, hipertermia, debilidad proximal y eritema en heliotropo. Fue manejado con tres bolos de metilprednisolona. Desarrolló eritema malar y vasculitis en pabellones auriculares, livedo reticularis, transaminasemia. Se indicó metotrexate 30 mg/m2SC/semana, prednisona 60 mg/día por 4 semanas y posterior reducción a 50 mg/día y además bloqueador solar. Electromiografía: parámetros normales, datos atípicos no concluyentes. Serie esófago-gastro-duodenal: leve retraso en el aclaramiento esofágico y en el vencimiento gástrico.
Los resultados del perfil enzimático y presencia de enfermedades autoinmunes se muestran en la Tabla 1.
Padecimiento actual. Once días de evolución con fiebre hasta 39 °C, un pico al día, sin predominio de horario, cedía parcialmente con antipirético; tos de tres días de evolución con paroxismos cortos, progresiva, disneizante, ciano-zante, expectoración blanquecina en moderada cantidad; dificultad respiratoria en las últimas 24 h, progresiva, con tiros intercostales, retracción xifoidea, cianosis peribucal y ungueal que se incrementaba con la tos.
Tratada en su lugar de origen con claritromi-cina 10 mg/kg/día, a su ingreso había tomado tres dosis.
Exploración física. Paciente masculino de 13 años de edad, con peso de 50 kg, talla 157 cm, FC 140/min, FR 58/min, TA 95/60 mmHg, temperatura 37.6 °C. Edad similar a la cronológica, sin facies característica, con cianosis peribucal y ungueal, normocéfalo, eritema en heliotropo bilateral, ojos simétricos, pupilas isocóricas, normorefléxicas, aleteo nasal, narinas permeables, membranas timpánicas íntegras, cavidad oral sin lesiones. Faringe con ligera hiperemia, cuello sin alteraciones, tráquea central, sin adenopatías, tórax normolíneo con tiros intercostales bajos, retracción xifoidea, campos pulmonares con hipoventilación basal bilateral. Ruidos cardiacos rítmicos, sin soplos. Abdomen blando, no doloroso, peristalsis normal, sin masas ni visceromegalias. Genitales masculinos Tanner 3. Extremidades íntegras simétricas, eutróficas. Neurológico con pares craneales normales, sin alteraciones sensitivas o motoras, fuerza global 5/5, reflejos osteotendinosos ++/++++, sin datos meníngeos.
Laboratorio. Hemoglobina (Hb) 15.8g/dL, hematocrito (Hto) 45.8%, leucocitos (Leuc) 19,000/ mm3, bandas (Ban) 8%, segmentados (Seg) 84%, plaquetas (Plaq) 332,000, glucosa (Gluc) 168 mg/dL, urea (BUN) 16mg/dL, creatinina (Creat) 1mg/dL, Na 139mEq/L, K 4.2mEq/L, Cl 96mEq/L, Ca 8.7mg/dL, P 4.1mg/dL, bilirrubina directa (BD) 0.1mg/dL, bilirrubina indirecta (BI) 0.31mg/ dL, transaminasa glutámico-oxalacético (TGO) 121U, transaminasa glutámico-pirúvico TGP 57U, deshidrogenas láctica (DHL) 1,021U, creatinina fosfocinasa (CPK) 20U, albúmina (Alb) 2.9g/L, proteínas totales (Prot T) 6.5g/L. Gasometría: pH 7.43, PaO2 51.8, PaCO2 25, HCO3 16.8, exceso de bases (EB) -5.4, SaO2 83%, lactato (Lact) 4.1. Examen general de orina (EGO): pH 6.5, densidad urinaria (DU) 1,010. Panel viral negativo, proteína C reactiva (PCReact) 3.17U.
RX tórax: infiltrado intersticial bilateral. Manejo: ayuno, soluciones de base 1,500mL/m2SC/ día 2:1, 30 mEq/m2SC/día de potasio, cefuroxima 150mg/kg/día, claritromicina 15mg/kg/día, FiO2 100% con mascarilla facial. Se suspendió metotrexate. USG abdominal: hepatomegalia con enfermedad hepática difusa; vesícula biliar, páncreas, riñones de apariencia normal. Debido a la inmunosupresión y la DHL elevada se sugirió descartar proceso por P. jiroveci.
5 de marzo-08. RX de tórax: patrón intersticial. Se agregó trimetroprim-sulfametoxasol (TMP/ SMX) 20mg/kg/día, se cambió cefuroxima por cepime y amikacina, continuó claritomicina. Inició BiPAP, sin mejoría. Ingresó a la terapia intensiva por dificultad respiratoria progresiva con intubación electiva, infusión de midazolam y fentanyl. Gasometría: pH 7.35, PaO2 60.6, PaCO2 47.5, HCO3 25.6, EB 0.6, SaO2 88%, Lact 1.1.
Continuó con deterioro ventilatorio por lo que se cambió a ventilación de alta frecuencia. TA hasta 80/50 mmHg, llenado capilar 3 seg. Taquicardia. Se indicó norepinefrina y reclutó en posición prona. Con disminución de FiO2 hasta 65%. Presentó fuga aérea y enfisema subcutáneo. Gasometría: pH 7.29, PaO2 73, PaCO2 54, HCO3 19.4, EB -3.9, SaO2 93%, Lact 3.9, Kirby 74.
8 de marzo 08. Inmunofluorescencia de broncoaspirado: negativa para Pneumocystis jiroveci. Cultivo de broncoaspirado para hongos: negativo. Se suspendió norepinefrina e hidrocortisona, se inició metilprednisolona a 4mg/kg/día. Se incrementó potasio a 40 mEq/m2SC/día. Continuó con hipotensión, fuga aérea y enfisema subcutáneo. Laboratorio: Hb 12.4g/dL, Hto 34.8%, Leuc 9,000/mm3, Ban 20%, Seg 76%, linfocitos (Linf) 2%, Plaq 99,000, Na 141mEq/L, K 3.2mEq/L, Ca 8.3mg/dL, P 2.8mg/dL, PCReact 7.8mg/dL.
12 de marzo 08. RX de tórax: infiltrado intersticial bilateral, neumomediastino y enfisema pulmonar. Se cambió a ventilación convencional, con presiones hasta 38 con hipoxemia, por lo que se reanudó ventilación de alta frecuencia, con Kirby de 60 a 80, Sat 92%, presión media 19, amplitud 23, FiO2 75%, Fr 3Hz, Ti 0.33.
15 de marzo 08. Se incrementó requerimiento ventilatorio. Se disminuyó metilprednisolona a 3mg/kg/día.
19 de marzo 08. Se agregó clonidina 5mg/kg/día y meropenem. Terminó 14 días con cefepime y TMP/SMX. Se disminuyó metilprednisolona a 1mg/kg/día. Laboratorio: Hb 10g/dL, Hto 29.5%, Leuc 8,200/mm3, Linf 2%, Ban 15%, Seg 80%, Plaq 157,000, Gluc 112mg/dL, BUN 9mg/dL, Creat 0.5mg/dL, Na 138mEq/L, K 4mEq/L, Cl 93mEq/L, Ca 8mg/dL, P 3mg/dL, BD 0.1mg/dL, BI 0.26mg/dL, TGO 84U, TGP 44U, Alb 2.7g/L, Prot T 6.3g/L. Hepatitis B y C negativos.
25 de marzo 08. Terminó 18 días con amikacina y claritromicina. Se intentó cambiar a ventilación convencional pero presentó progresión de la fuga aérea, cianosis central, hipotensión y se reinició ventilación de alta frecuencia. RX de tórax: neumotórax derecho; se colocó sello pleural con mejoría. Gasometría: pH 7.36, PaO2 48, PaCO2 78, HCO3 43, EB -4.9, SaO2 79%, Lact 1.8.
26 de marzo 08. Ag Candida positivo. Formas invasivas orina Candida tropicalis. Se agregó anfotericina B e insulina.
Laboratorio: Hb 9g/dL, Hto 27.1%, Leuc 15,300/mm3, Linf 1%, Ban 13%, Seg 80%, Plaq 253,000, Gluc 238mg/dL, BUN 14mg/dL, Creat 0.5mg/dL, Na 137mEq/L, K 3.3mEq/L, TGO 8,419U, TGP 7,806U, Amilasa 108U, Lipasa 220U, DHL 7,439U, fosfatasa alcalina (FA) 111U, PCReact 4.93mg/dL.
31 de marzo 08. Hb 7.5g/dL. Presentó taquicardia, baja de la reserva venosa de 67 a 42%, lactato de 2 a 3.5. Se manejó con norepinefrina y concentrado eritrocitario 15mL/kg/dosis.
1° de abril 08. Continuó con apoyo aminérgico e incremento progresivo de lactato. FC hasta 30/min, TA media hasta 55 mmHg, sin respuesta a aminas. Kirby máximo 40, índice de oxigenación (IO) hasta 100, Sat hasta 60%. Presentó enfisema subcutáneo extenso, sello pleural con fístula bronqueal, datos de fibrosis y necrosis pulmonar, fuga aérea progresiva y paro cardiorrespiratorio irreversible.
Discusión clínica
Dra. Floricela Olivera Solórzano. Se trató de un paciente masculino con antecedente de importancia de una hermana de 11 años de edad estudiada en otra unidad con enfermedad autoinmune. Fue referido a nuestra institución por cuadro de un mes de evolución debido a lesiones dérmicas caracterizadas por eritema en heliotropo, debilidad muscular proximal en las cuatro extremidades e infección de vías aéreas superiores. Se le diagnosticó dermatomiositis juvenil probable en base a los criterios de Bohan y Meter propuestos en 1975 y que consisten en: 1) debilidad muscular proximal y simétrica en las cinturas escapular y pélvica; 2) elevación de las enzimas musculares 20 a 40 veces su valor normal; 3) patrón miopático en la electromiografía; 4) biopsia muscular compatible con miopatía inflamatoria y 5) lesiones dérmicas características.1 Se hace el diagnóstico definitivo cuando se tienen más de tres criterios con lesiones cutáneas características; de probabilidad, cuando se tienen dos criterios más las lesiones cutáneas; y de posibilidad, cuando hay un solo criterio más las lesiones de la piel. Los anticuerpos antinucleares (ANA) no son específicos en la dermatomiositis y se encuentran con una frecuencia variable de 10 a 85%. Hay auto anticuerpos específicos como anti-sintetasa (anti-aminoacetil-tRNA sintetasa) descritos como antisintetasas Jo-1, PL-7, PL-12, anti Mi-2 y anti SRP. También existen anticuerpos asociados a miositis como Anti-PM-Scl, Anti Ku, Anti-anexin XI, anti-U y anti-SSA/Ro. Estos anticuerpos no tienen valor como criterio diagnóstico y se relacionan con valor pronóstico. En el paciente, el anti Jo-1 y los anti Sm fueron negativos aunque su sensibilidad es baja ya que solo se encuentran en 20-30% de los pacientes. En la actualidad, la biopsia muscular y la electromiografía están en desuso por ser procedimientos invasivos, dolorosos e innecesarios si se cumplen los criterios de afección dérmica, debilidad muscular y elevación de enzimas musculares. La elevación de CPK y DHL son más específicas pero la TGO y TGP también se correlacionan. En sustitución de la biopsia muscular se puede utilizar la resonancia magnética nuclear con extracción de grasa, la cual muestra los paquetes musculares con los sitios de inflamación de tal forma que, si fuese necesario, la biopsia se tomaría de los sitios adecuados.2 En el paciente se consideró el diagnóstico como probable por las lesiones cutáneas, la debilidad muscular y enzimas elevadas. A su llegada a nuestro hospital sólo persistían las lesiones dérmicas y la debilidad muscular, las enzimas estaban normales y la electromiografía fue no concluyente. Ante ello se puede considerar la dermatomiositis amiopática.3 La dermatomiositis juvenil se clasifica en la forma clásica cuando hay alteraciones musculares y en piel, amiopática cuando sólo hay alteraciones dérmicas y la forma hipoamiotrófica con espectro entre ambos con discreta debilidad muscular o leve elevación de enzimas. En el hospital de referencia se dio tratamiento con pulsos de metrilprednisolona y se inició prednisona y me-totrexate. Para el tratamiento en la fase aguda del padecimiento se recomienda prednisona a dosis de 60mg/kg/día y el metotrexate a 15-20mg/m2/ día como medicamento de base. Dosis mayores de 15-20mg/día no son mejores y hay riesgo de toxicidad.4 En los casos en que los hay dificultad respiratoria por afección de los músculos respiratorios o reflujo gastroesofágico que pone en riesgo la vida del paciente, se recomienda gammaglobulina y en los casos refractarios rituximab o infliximab, entre otros. La electromiografía reportó un patrón no concluyente y la serie esófago-gastro-dudenal leve retraso en el vaciamiento gástrico. Este último estudio es importante ya que, en el paciente pediátrico, la afección esofágica es de mal pronóstico y el reflujo gastroesofágico puede condicionar pneumonitis por aspiración. En el diagnóstico diferencial se deben considerar otras miositis inflamatorias, como las infecciosas por influenza tipo A, B, adenovirus, y parasitarias como triquinosis, tripanosoma y toxoplasma. También deben considerarse las distrofias musculares de Duchenne y Becker y la miositis por inclusión. La evolución del paciente fue mala con tos, dificultad respiratoria y fiebre compatible con un cuadro infeccioso de vías aéreas. Se le dio tratamiento con claritromicina a dosis de 10mg/kg/día, la cual es una dosis inadecuada y progresó a respuesta inflamatoria sistémica con DHL elevada. El deterioro progresivo fue mayor y requirió de ventilación mecánica asistida con parámetros altos y escalando a ventilación de alta frecuencia con desarrollo de neumotórax y enfisema subcutáneo. En los pacientes inmunosuprimidos y con enfermedades reumatológicas se deben sospechar agentes virales como herpes, citomegalovirus, virus sincicial respiratorio, influenza, etc.; bacterias como E. coli, Salmonella, Klebsiella, H influenza, meningitides, Enterobacter, entre otros. Ante la DHL elevada, en Pneumocystis jirovecci y sin dejar de pensar en tuberculosis o infección por hongos. En conclusión, nuestro paciente cursó persistentemente con hipoxemia, acidosis respiratoria severa, hipotensión y bradicardia, requirió ventilación de alta frecuencia con daño a la vía aérea, con fuga caracterizada por neumotórax y enfisema que agravaron aún más el daño.
Diagnósticos clínicos finales. Adolescente masculino eutrófico, dermatomiositis juvenil, síndrome de fuga aérea con enfisema y neumotórax, neumonía nosocomial y choque séptico.
Hallazgos anatomopatológicos
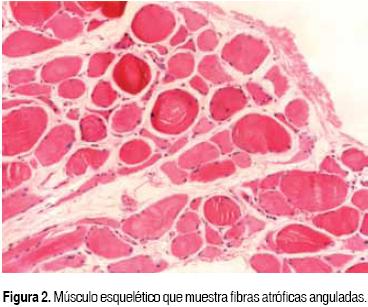
Dr. Guillermo Ramón García (Médico adscrito Departamento de Patología). Al momento de la autopsia, el paciente aún presentaba lesiones dérmicas en la cara como son el eritema facial alrededor de las mejillas y alrededor de los ojos. Se tomó un fragmento de piel que muestra en la dermis, alrededor de los vasos, leve infiltrado inflamatorio linfohistiocitario perivascular (Figura 1). Se tomaron también músculos en diversos sitios que mostraron fibras atróficas, pequeñas, anguladas, situadas a la periferia de los fascículos. No se encontró inflamación o necrosis en ninguno de los músculos muestreados (Figura 2). La combinación de la lesión inflamatoria en dermis con la atrofia perifascicular en los músculos apoyan el diagnóstico de dermatomiositis.5 Como se puede desprender del análisis de la historia clínica, las complicaciones más importantes en este paciente estuvieron en el aparato respiratorio. En esta imagen se muestran los pulmones al corte, cuya mucosa de tráquea y bronquios presenta necrosis extensa. Los ganglios peritraqueales y peribronquiales están aumentados de tamaño (Figura 3). Los pulmones tienen aspecto sólido en múltiples áreas alternando con zonas hemorrágicas. Se encontró neumotórax residual en el lado derecho. Microscópicamente se corrobora la necrosis extensa en toda la mucosa de las vías aéreas que se extiende a toda la pared. El parénquima pulmonar muestra alveolos ocupados por edema, fibrina, membranas hialinas que corresponden a daño alveolar difuso agudo, y zonas de necrosis e inflamación aguda múltiples que corresponden a áreas con neumonía de focos múltiples (Figura 4). Los ganglios linfáticos muestran hiperplasia de senos con numerosos histiocitos con hemofagocitosis (Figura 5). Esta hemofagocitosis también fue observada en la médula ósea, hígado y bazo. El hígado estaba congestivo, aumentado de peso y, microscópicamente, presentaba esteatosis difusa de gota gruesa. En el páncreas había esteatone-crosis y leve infiltrado inflamatorio. Los riñones estaban congestivos y aumentados de peso; microscópicamente se observó, en los túmulos, edema acentuado de su epitelio con cambios de degeneración hidrópica. En el encéfalo se encontró hemorragia subaracnoidea focal sobre los hemisferios a nivel de los lóbulos occipitales, acompañado de edema y congestión; al corte se aprecian pequeños infartos sobre la región occipital y frontal.




Los diagnósticos finales son:
Enfermedad principal:
• Dermatomiositis
Alteraciones concomitantes:
• Neumonía intersticial aguda
• Laringotraqueobronquitis necrosante ulcerada
• Neumotórax residual
• Hepatomegalia con esteatosis difusa
• Nefromegalia con nefrosis osmótica
• Edema cerebral
• Hemorragia subaracnoidea occipital
• Infartos en lóbulo frontal y occipital
• Síndrome de hemofagocitosis secundaria
Causa de la muerte:
• Daño alveolar difuso
Comentarios finales
Dr. Adrián Chávez (Jefe de Terapia Intensiva).
Se trató de un paciente con un diagnóstico poco frecuente que tuvo múltiples complicaciones. La principal fue la neumonía intersticial aguda de origen viral, que probablemente le condujo a insuficiencia respiratoria y que motivó que se utilizara ventilación de alta frecuencia complicándose a su vez con fuga aérea. La lesión necrosante de la mucosa laringotraqueal es característica de este tipo de ventilación. Con una lesión pulmonar grave, con Kirby menor de 100 como la que se presentó en este paciente, considero debió iniciarse con ventilación de alta frecuencia y no de moda de rescate. En este pulmón con fuga era muy difícil lograr una ventilación y oxigenación adecuadas.
Dra. Rocío Maldonado (Médico adscrito a Reumatología). Con respecto al síndrome de activación de macrófagos, ésta es una entidad que se encuentra en múltiples enfermedades, como la linfohistiocitosis familar asociada a infecciones o en enfermedades reumatológicas en las que hay una alteración de todo el sistema inmune caracterizada por activación de macrófagos y en la que se observa fagocitosis extensa que se comporta agresivamente y que puede conducir a la muerte del paciente. Es poco común en la dermatomiositis; es más frecuente en la artritis reumatoide (AR) y en lupus eritematoso sistémico (LES).6 En este caso en particular, considero que la activación de macrófagos fue secundaria a la neumonía de origen viral. Para hacer este diagnóstico debieron haberse determinado los niveles de ferritina y otros parámetros, pero no se hicieron con en este paciente.7
Referencias
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med 1975;292:344–347;403-407. [ Links ]
2. Babyn P, Doria AS. Radiologic investigation of rheumatic diseases. Rheum Dis Clin North Am 2007;33:403–440. [ Links ]
3. Gerami P, Walling HW, Lewis J, Doughty L, Sontheimer RD. A systematic review of juvenile-onset clinically amyo-pathic dermatomyositis. Br J Dermatol 2007;157:637–644. [ Links ]
4. Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet 2008;371:2201–2212. [ Links ]
5. Anthony DC, Frosch MP, DeGirolami U. Enfermedades del músculo-esquelético. In: Kumar V, Abbas AK, Fausto N, eds. Robbins y Cotran Patología Estructural y Funcional. España: Elsevier; 2005. p. 1347. [ Links ]
6. Grom AA. Natural killer cell dysfunction: a common pathway in systemic-onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis? Arthritis Rheum 2004;50:689-698. [ Links ]
7. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol 1991;18:29-33. [ Links ]

