










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versão impressa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.67 no.4 México Jul./Ago. 2010
Caso clínico patológico
Lactante con cuprotoxicosis idiopática
Idiopathic copper toxicosis in a 1-year-old male
María de Lourdes Cabrera-Muñoz1, José Domingo Gamboa-Marrufo2, Olga Martínez Pantaleón3, Pedro Bazán Santos4
1 Departamento de Patología
2 Subdirección de Atención Integral al Paciente
3 Departamento de Urgencias
4 Departamento de Radiología; Hospital Infantil de México Federico Gómez, México, D.F., México
Autor de correspondencia:
Dra. Ma de Lourdes Cabrera Muñoz
Correo electrónico: cabreramalu@aol.com
Fecha de recepción: 04-02-10.
Fecha de aceptación: 24-02-10.
Introducción
El cobre es un elemento traza esencial para algunos de los procesos biológicos de plantas y animales. Sirve como cofactor de grupos prostéticos en la función de algunas enzimas y es responsable de la expresión de genes específicos.1
Las concentraciones del cobre en las plantas y animales son influenciadas por las condiciones locales, particularmente por el contenido del cobre en la tierra y el agua.
Algunos alimentos como vísceras (hígado, riñón) y algunos frutos y nueces tienen alto contenido de cobre.2
La acumulación de cobre en el hígado ha sido observada en una variedad de enfermedades hepáticas pediátricas que incluyen: enfermedad de Wilson,3 cirrosis del niño indio,4 toxicosis idiopática por cobre;5 estas dos últimas han sido relacionados con dietas ricas en cobre. La toxicosis idiopática por cobre (TIC) aparece esporádicamente fuera del continente indio; se han reportado 15 casos por década en las últimas décadas.6 Existe un cuadro clínico que es indistinguible de ésta, que se llama cirrosis infantil endémica del tirol.7 En México, este cuadro clínico ya ha sido reportado por Valencia y Gamboa, en 1993.8
Al no existir muchos de casos, se debe tener presente la existencia de esta entidad, y en cualquier paciente con colestasis, anemia y desnutrición se deberá contar con la cuantificación de cobre en sangre y orina. Sin embargo, se debe recordar que las concentraciones de cobre y ceruloplasmina pueden ser normales o estar ligeramente aumentadas.
Resumen de la historia clínica
Masculino de un año que inicia su padecimiento actual a los dos meses de vida con ictericia, que ha sido progresiva y generalizada. Distensión abdominal desde los dos meses y medio, insidiosa, no progresiva y aumento del volumen escrotal al mismo tiempo, no progresivo. Se niega hipoacolia.
Exploración física:
A la exploración física encontramos los siguientes datos: peso 6,085 g, talla 58 cm, F.C. 160/min, F.R. 32/min, T.A 90/60 mm/Hg, temperatura 37.3ºC, llenado capilar 2 segundos, perímetro cefálico 41 cm, perímetro abdominal 46 cm.
Edad aparente menor a la cronológica, facies tosca, abotagada, ictericia generalizada, cabello seco y frágil. Edema bipalpebral, puente nasal ancho, implantación baja de pabellones auriculares, tórax restrictivo por distensión abdominal, soplo sistólico en tercer espacio intercostal izquierdo grado II/VI. Abdomen globoso a expensas de líquido de ascitis, hígado a 9-8-8 cm abajo del borde costal y bazo a 7 cm. Hernia umbilical de 2 cm, reductible. Extremidades edematizadas. Godete positivo, acropaquia leve. Neurológico normal, reflejos O.T. ++/++++, petequias en región sacrococcígea.
Exámenes de laboratorio
Hb 10.6 g/dL, plaquetas 15000/mm3, leucocitos 27600/mm3, Seg 74%, lin 18%, Mon 4%, Bon 2%, glucosa 64 mg/dL, Ca 7.5 mg/dL, P 2.5 mg/dl, amonio 105 mmol/L.
TP 28" (T 12"), TTP 54.1" (T26"), INR 2.21, BD 24.6 mg/dL, BI 3.92 mg/dL.
Prot totales 6.2 g/dL, Alb 2.1 g/dL, Glob 4.1 g/ dL, TGO 383 UI, TGP 384 UI, GGT 43 U/L, FA 84 U/L, DHL 406 U/L.
EGO: pH 8.0, DU 1010, Hb 10 erit//mL, bilis 6 mg/dL, calcio urinario 7.7 mg%, vol. 280 mL. Gasometría pH 7.41, PaO2 45, Pa CO2 32.9, HCO3 23.6, CO2T 28.9, EB-0.4, SaO2 71.2, Lactato 2.9
Es visto por endocrinología y se hace diagnóstico de hipotiroidismo; se inicia manejo con levotiroxina 6.2 μg/kg/día. Cardiología lo considera normal.
USG abdomen, líquido libre en cavidad abdominal, nefrocalcinosis bilateral.
USG hepático Doppler. Vena porta con flujo turbulento, velocidad 10.3 cm/seg en porción principal, 14.7 cm/seg rama derecha y 21 cm/ seg rama izquierda.
Índice de resistencia (IR) arterial hepática: 0.85, glucosa capilar 39 mg/dL.
Biopsia hepática por trucut bajo visión endoscópica, plastía inguinal bilateral, plastía umbilical e hidrocelectomía bilateral. Hígado color verde-morado oscuro con superficie nodular y red verdosa superficial, la biopsia hepática mostró parénquima hepático con arquitectura perdida, numerosos linfocitos y proliferación de colangiolos, los espacios porta se encuentran ensanchados, con fibrosis y puentes necroinflamatorios, no se observan venas centrales; los hepatocitos presentan degeneración hidrópica, son de citoplasma claro, con numerosos cuerpos de Mallory, colestasis intra y extra celular con formación de tapones de bilis en los colangiolos; estos se encuentran proliferando y acompañan los puentes necroinflamatorios, los núcleos muestran pleomorfismo leve. Los cam-bios son compatibles con toxicosis idiopática por cobre.
El paciente evoluciona a insuficiencia renal, deterioro ventilatorio y fallece. Estuvo del 3 al 21 de julio de 2008 hospitalizado.
Hallazgos radiológicos
En el ultrasonido se observa hepatomegalia de ecogenicidad homogénea, no hay dilatación de vías biliares intra ni extrahepáticas (Figura 1).
Con la señal Doppler a nivel del hilio hepático, la porta se observa con velocidad de 10 cm/seg. A nivel del hilio esplénico muestra 24 cm/seg. La arteria hepática con IR de 0.85 (Figura 2).
En el ultrasonido renal, los riñones se encuentran discretamente aumentados de tamaño, las pirámides que normalmente son hipoecoicas se observan hiperecoicas a expensas de calcificaciones de bordes mal limitados compatibles con nefrocalcinosis (Figura 3).
Las venas suprahepáticas dentro de límites normales. En las placas del tórax y huesos largos llama la atención el aumento del volumen y la densidad de los tejidos blandos.
Discusión
Lactante masculino de 12 meses de edad; dentro de sus antecedentes personales patológicos tenemos que fue un recién nacido pretérmino moderado con peso bajo para la edad de gestación de acuerdo a las gráficas adaptadas de Babson (32.4 SDG con peso de 1300 g –se encuentra en la percentila 2). Es obtenido por parto y se refiere sin llanto ni respiración espontáneos, lo que sugiere que cursó con probable asfixia perinatal. Es importante señalar que este paciente debió ser obtenido por cesárea si es que se estaba desencadenando el trabajo de parto, para evitar la asfixia y trauma obstétrico.
Se refiere además sepsis e hiperbilirrubinemia en apariencia multifactorial, no se especifican niveles de bilirrubinas ni manejo, cursó con hemorragia intraventricular; todos estos eventos lo hacen portador de alto riesgo neurológico, por lo que se envía a esta unidad a los 4 meses de edad. A los 8 meses de edad se tiene un ultrasonido transfontanelar con atrofia cortical y ventriculomegalia, seguramente como secuelas de la asfixia perinatal y de la hemorragia intraventricular y a los 11/12 meses presenta hipotonía central; los hallazgos se atribuyen a secuelas de encefalopatía bilirrubinémica crónica.
El cuestionamiento que sigue es: ¿por qué fue un paciente pretérmino? Se menciona en la historia clínica que aparentemente no hubo factores maternos, como infección materna ni hipertensión; sin embargo, no se cuenta con una historia detallada del embarazo ni exámenes de laboratorio. Entre los factores que destacan para desencadenar un parto pretérmino son: sexo masculino, factores ambientales que incluyen la pobreza, escasos recursos para cuidados básicos y educación, como ha sido comentado por el personal del departamento de Trabajo Social.
A la edad de 12 meses, cuando ingresa para estudio integral, el paciente tiene una desnutrición grave, de acuerdo a indicadores antropométricos según la clasificación del Dr. Gómez, con un déficit de peso del 58% respecto al esperado para su edad; por la descripción a su ingreso (con edema de extremidades) probablemente se trate de una desnutrición tipo Kwashiorkor, y de acuerdo a la clasificación de Waterlow se catalogaría como desnutrición crónica agudizada.
El paciente cursa con retraso severo en el desarrollo psicomotor. A este respecto existen en el paciente factores de riesgo que lo explican, como son: factores biológicos (peso al nacer menor de 1,500 g, edad gestacional de menos o igual a 32 semanas, asistencia ventilatoria por más de 36 h, hemorragia intraventricular) y factores de riesgo establecido (asfixia perinatal), así como factores ambientales ya mencionados previamente e hipotiroidismo congénito fundamentado en pobre incremento ponderal y un fenotipo compatible con facies tosca, piel seca, fontanela anterior amplia y hernia umbilical. Las pruebas de función tiroidea apoyan el diagnóstico clínico y permiten sospechar de hipotiroidismo congénito secundario. Este último es un diagnóstico tardío debido a que a los 6 meses de edad el hipotiroidismo no tratado reduce el IQ en 50%. Considero importante mencionar que, en México, en la mayoría de los casos, el hipotiroidismo congénito es primario; la frecuencia es de 1:2537 casos (alta en relación a otros países) y, debido a que al nacimiento sólo menos de 5% presentan datos clínicos, se recomienda que se realice el tamiz neonatal en todos los recién nacidos.
El paciente cursa con ictericia desde los 2 meses de edad, con hiperbilirrubinemia a expensas de bilirrubina conjugada, por lo que se agrega el diagnóstico de síndrome colestásico; también se agrega a los diagnósticos sindromáticos hepatoesplenomegalia, documentada clínicamente y por imagen, así como hipertensión porta y ascitis de curso crónico.
Síndrome colestásico
Las causas son variadas y estamos obligados a descartar las siguientes:
1. Etiología Infecciosa. La lesión hepatocelular secundaria a un proceso infeccioso no se descarta. El paciente cuenta con serología negativa para hepatitis A, B, EBV, VDRL; sin embargo, falta la determinación para citomegalovirus, rubéola, herpes, virus de inmunodeficiencia y toxoplasmosis.
2. Obstrucción de vías biliares. Lo obligado en estos casos es descartar atresia de vías biliares. Esta entidad se presenta en 1:8,000 casos, consiste en atresia o hipoplasia de cualquier porción del sistema biliar extrahepático; por lo regular, son niños de término con peso adecuado para la edad gestacional que presentan ictericia a las 2-3 semanas de edad, acompañada de acolia; el ultrasonido abdominal, donde no se observa la vesícula biliar, apoya esta posibilidad. Sin embargo, este paciente no presentó acolia y la biopsia hepática no es compatible. Habría sido útil contar con un gammagrama hepático donde se podría observar falta de excreción del material radioactivo al intestino y una colangiografía transoperatoria. No puedo excluir totalmente a la atresia de vías biliares. El síndrome de Alagille presenta pérdida de conductos biliares intrahepáticos, se caracteriza por facies anormal (frente amplia, hipertelorismo, nariz larga-recta con ala de la nariz ancha y micrognatia), cardiopatía tipo estenosis pulmonar o tetralogía de Fallot, anomalías en columna vertebral (vértebras en mariposa, hemivertebras), alteraciones oftalmológicas (embriotoxón) retardo en el crecimiento y retraso mental. Por no contar con estos hallazgos, esta posibilidad queda descartada.
3. Enfermedades congénitas del metabolismo. Se menciona que en el hombre se conocen más de 2000 enzimas distintas, por lo que deberían de existir al menos 2000 enfermedades congénitas del metabolismo, aunque sólo se conocen aproximadamente 400. En estos casos es de utilidad el tamiz metabólico ampliado (carecemos de este estudio, al menos en la historia clínica). Mencionaré las compatibles con el cuadro clínico:
a) Tirosinemia. Es un desorden multisistémico progresivo que afecta al hígado, riñón y sistema nervioso periférico. La disfunción hepática progresiva inicia en la infancia y, en edades más avanzadas, se manifiesta el cuadro clínico característico; sin embargo, en algunos pacientes, la enfermedad es de un curso rápidamente progresivo y desarrollan falla hepática al año de edad. Es característico también un grado variable de disfunción renal (acidosis tubular renal, glomeruloesclerosis e insuficiencia renal) y la presencia de nefrocalcinosis en el 33% de los casos. El paciente, a pesar de tener la afectación hepática, no presenta datos de acidosis tubular renal y la función renal a su ingreso es normal. El diagnóstico se apoya con los niveles de tirosina, pero queda como posibilidad remota.
b) Esfingolipidosis (incluyendo enfermedad de Gaucher-Colman y Nieman Pick). Cursan con fibrosis hepática. Lo llamativo, además, es la hipercolesterolemia y aumento de los triglicéridos, situación que no correlaciona con nuestro paciente. La biopsia revela lípidos intralisosomales, por lo que se descarta esta etiología.
c) Glucogenosis Tipo I. Se presenta hepatomegalia con acidosis láctica e hipoglicemia, lo que no se confirma en el paciente a su ingreso; el paciente presenta hiperlactatemia e hipoglicemia en el curso de enfermedad grave, que correlaciona más con el estado de deterioro hemodinámico y el desarrollo de la insuficiencia hepática que presentó en el estadio final de la enfermedad.
d) Enfermedad de Wilson. Es un desorden autosómico recesivo del metabolismo del cobre; se reporta que es raro en pacientes menores de 5 años de edad. El Dr. Fontana, de la Universidad de Michigan, hizo recientemente énfasis en considerar siempre esta posibilidad en casos de hepatoesplenomegalia acompañada de insuficiencia hepática de evolución catastrófica y cuya causa no esté bien establecida. En la enfermedad de Wilson también existen otros sitios de toxicidad por acumulación de cobre como son sistema nervioso central, ojos y riñones. En nuestro paciente, la disfunción de la hipofisiaria pudo deberse a la acumulación de cobre. La nefrocalcinosis es un hallazgo descrito en esta enfermedad. Habría sido de ayuda contar con una resonancia magnética del cerebro. En el examen físico no se describen anillos de Kaiser-Fleisher, pero estos pueden estar ausentes en el 50% de los casos. El diagnóstico se realiza con la determinación de cerulopasmina sérica baja, nivel de cobre en sangre alto y cobre en orina; en la biopsia hepática con tinciones especiales se puede detectar los depósitos de cobre a nivel celular y cuantificar la concentración de cobre por gramo de tejido hepático. No puedo descartar esta enfermedad ya que no cuento con estudios de laboratorio.
e) Enfermedad por atesoramiento de hierro. Me refiero a la hemocromatosis neonatal, donde el hierro se deposita en hígado, páncreas, piel, sistema endocrino (tiroides). En esta enfermedad hay historia de prematurez, retardo en el crecimiento intrauterino, el cuadro clínico de hepatitis con hepatomegalia y síndrome colestásico; puede presentarse en las primeras semanas de vida y, en la mayoría de los casos, evoluciona como una hepatitis fulminante. El diagnóstico se confirma con: hierro en plasma aumentado, transferrina disminuida, saturación de transferrina aumentada, ferritina aumentada. La biopsia hepática también es de ayuda al diagnóstico ya que con tinciones especiales se pueden detectar los depósitos de hierro, así como la cuantificación de hierro por gramo de tejido hepático. Esta causa no queda descartada.
4. Entre las causas misceláneas se encuentra la hepatotoxicidad por xenobióticos: por medicamentos (paracetamol), metales (ingesta aguda de sulfato de cobre, de hierro), hongos de la especie amanita (necrosis hepática) y herbolaria (derivados pirrolizínicos que producen enfermedad veno-oclusiva , gordolobo). También fibrosis quística y alimentación parenteral prolongada.
Otros diagnósticos
Insuficiencia hepática. El paciente cursó con hi poalbuminemia, alteración de la coagulación con INR mayor de 1.5 con aumento de las bilirrubinas e hipoglicemia.
Proceso infeccioso relacionado a neumonía nosocomial.
Evolución
Se realiza biopsia hepática con deterioro de su estado clínico. Presenta infección nosocomial, alteraciones metabólicas y electrolíticas, datos de daño pulmonar agudo y de choque, con falla orgánica múltiple. Llama la atención el descenso constante de hemoglobina, a pesar de las trans fusiones lo que, aunado a las alteraciones de la coagulación, requería de sospechar pérdidas de sangre o hemólisis.
Diagnósticos finales
Lactante masculino de un año de edad, desnutrición crónica agudizada, retraso psicomotor, hipotiroidismo congénito secundario, status post biopsia, status post hernioplastías- hidrocele, hepatoesplenomegalia, hipertensión porta, síndrome colestásico, neumonía nosocomial, insuficiencia hepática y status post paro.
Causa de muerte
Falla orgánica múltiple y choque mixto.
Descripción de los hallazgos histopatológicos
La biopsia hepática que se le realizó al paciente muestra hepatocitos con colestasis, transformación pseudoacinar y citoplasma claros, donde se observan numerosos cuerpos hialinos de Mallory y fibrosis intersticial. En el estudio de ultraestructura, las mitocondrias de los hepatocitos no presentan alteraciones morfológicas (Figura 4). Con estos hallazgos se realizó el diagnóstico compatible con cuprotoxicosis.
En la autopsia, el paciente muestra ictericia generalizada, distensión abdominal, red venosa colateral, hernia umbilical, hidrocele bilateral y en los ojos se observa coloración oscura en el limbo esclerocorneal que corresponde a anillos de Kayser- Fleischer (Figura 5). En los niños se han descrito anillos de Kayser-Fleischer en varias enfermedades que cursan con colestasis (Tabla 1) y son debidos al depósito de cobre en la membrana de Descement.

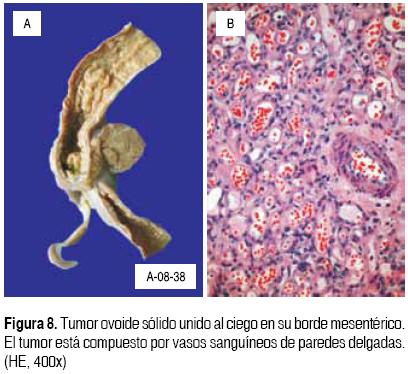
El hígado está aumentado de tamaño y peso, su superficie externa y de corte es micronodular, verdosa y los nódulos miden menos de 3 mm. Histológicamente se observan nódulos de regeneración rodeados por tabiques de tejido conectivo, con proliferación de colangiolos. En los nódulos existe fibrosis intersticial y los hepatocitos muestran numerosos cuerpos hialinos de Mallory, colestasis y degeneración de hepatocitos con satelitosis por polimorfonucleares. En las reacciones de inmunohistoquímica, los cuerpos de Mallory son positivos para citoqueratinas de bajo y alto peso molecular y la intensidad de la tinción es mayor para citoqueratina de alto peso molecular (Figura 6). No se cuenta con tinción de histoquímica para demostrar cobre, por lo que se realizó tinción con orceína para demostrar el depósito de la proteína asociada a cobre, la cual es positiva en el citoplasma de los hepatocitos. Los riñones están aumentados de tamaño y presentan datos de necrosis tubular aguda y cilindros de pigmentos biliares. El estudio microscópico del tubo digestivo solamente muestra cambios de miopatía visceral hipóxicoisquémica secundaria a choque, y los pulmones con una neumonía aguda en fase de resolución. En la médula ósea, los precursores de las tres series no presentan alteraciones y existe osteopenia leve. El timo es pequeño y atrófico. El cerebro muestra atrofia subcortical y dilatación de los ventrículos. Los diferentes cortes histológicos de la corteza y núcleos grises se encuentran con cambios crónicos, caracterizados por pérdida neuronal y encefalopatía metabólica, así como necrosis multifocal de neuronas reciente (Figura 7). Las malformaciones congénitas que se encontraron en este paciente son un cavum septum pellucidum, comunicación interauricular tipo foramen oval de 2 mm, y hemangioma capilar en mesocolon de ciego (Figura 8). Estas últimas no fueron detectadas clínicamente.
Las enfermedades metabólicas que presentan un patrón morfológico de cirrosis hepática incluyen a la enfermedad de Wilson y a la cuprotoxicosis, entre otras (Tabla 2).9 Los cambios morfológicos pueden ser indistinguibles en las fases iniciales; sin embargo, existen algunas diferencias que hacen posible su separación cuando se analizan las biopsias. Los datos más importantes son la edad a la presentación de los síntomas, el antecedente familiar o el antecedente de la exposición a utensilios de cobre. También se recomienda el estudio molecular.

La experiencia en el Hospital Infantil de México Federico Gómez de casos de cirrosis asociados a toxicidad por cobre fue publicada por Valencia y Gamboa (Tabla 3).8 Los cuatro pacientes son del género masculino y tres de ellos son menores de dos años, como el paciente que aquí se presenta.

Los diagnósticos finales son los siguientes:
Enfermedad principal
Cuprotoxicosis idiopática
Alteraciones concomitantes
Cirrosis hepática micronodular
Colestasis grave
Hepatomegalia 300 g vs 280 g
Nefropatía por bilirrubina
Datos de hipertensión portal
Neumonía aguda en fase de resolución
Atrofia del timo
Atrofia cerebral con dilatación de ventrículos cerebrales
Encefalopatía metabólica
Edema cerebral 900 g vs 852 g
Encefalopatía hipóxica aguda
Alteraciones independientes
Hipotiroidismo
Hernia umbilical
Status post plastía inguinal bilateral
Hemangioma capilar en mesocolon
Comunicación interauricular tipo foramen oval 2 mm
Cavum septum pellucidum
La edad de inicio de los síntomas de la toxicosis idiopática por cobre (TIC), descrita en la literatura, varía de los 2 meses a los 10 años, como es el caso de nuestro paciente. Tres grupos de inicio pueden ser distinguidos: infantil, si inició antes o alrededor de los dos años, inicio tardío, alrededor de los 5 años y retraso excepcional, alrededor de los 10 años.10-12
La cirrosis del niño indio se presenta entre los 6 y 18 meses13 y la sintomatología de la enfermedad de Wilson inicia alrededor de los 6 años.14
En la mayoría de los casos, la TIC está caracterizada como una enfermedad de inicio insidiosa, de rápida progresión y muerte en el primer año, como es el caso de nuestro paciente. En ocasiones tiene una presentación crónica que dura varios años y termina como una insuficiencia hepática.
Los exámenes de laboratorio que nos permiten hacer el diagnóstico son las concentraciones altas de cobre en sangre e hígado (entre 190 y 360 μg/g de hígado seco); la concentración normal es menor de 50 μg/g.8,10,11,14 Se ha observado también aumento de la excreción de cobre por orina. La ceruloplasmina por lo general es normal o ligeramente elevada.
El estudio que confirma este diagnóstico no es sólo la biopsia hepática, sino la cantidad de cobre que se haya almacenado en el hígado.
El factor etiológico principal de esta enfermedad es el uso constante de utensilios hechos de cobre, ya que se pueden contaminar los alimentos, como en el caso de la leche, y pueden producir acumulación de cobre y la producción de la enfermedad.7 En nuestro país existen sitios como Santa Clara del Cobre, en el estado de Michoacán, donde fabrican utensilios para cocinar de este material. Aunque, al parecer, no es muy frecuente esta enfermedad, se debe tener presente, ya que puede ser tratada tempranamente con D-penicilamina11,12 y también se puede efectuar trasplante hepático.
Referencias
1. Lincer MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr 1996;63:5797-5811. [ Links ]
2. Pandit AN, Bhave S. Copper. Metabolic defects and liver disease: environmental aspects. J Gastr Hepat 2002;17:403-407. [ Links ]
3. Gollan JL, Gollan TJ. Wilson disease in 1998: genetic, diagnostic and therapeutic aspects. J Hepatol 1998;1:28-36. [ Links ]
4. Tanner MS. Role of copper in Indian childhood cirrhosis. Am J Clin Nutr 1998;67:1074-1081. [ Links ]
5. Muller T, Muller W, Feichtinger H. Idiopathic copper toxicosis. Am J Clin Nutr 1998;67:1082-1086. [ Links ]
6. Muller T, Schafer H, Rodeck B, Haupt G, Koeb H, Bosse H, et al. Familial clustering of infantile cirrhosis in Northern Germany: a clue to the etiology of idiopathic copper toxicosis. J Pediatr 1999;135:189-196. [ Links ]
7. Muller T, Feichtinger H, Berger H, Muller W. Endemic tyrolean infantile cirrhosis: an ecogenic disorder. Lancet 1996;347:877-880. [ Links ]
8. Valencia MP, Gamboa MJ, Medina J. Copper overload and cirrhosis in four Mexican children. Lab Invest 1993;68:10 P(abstr). [ Links ]
9. Jevon GP, Dimmick JE. Histopathological approach to metabolic liver disease. Part 1. Pediatr Develop Pathol 1998;1:261. [ Links ]
10. Baker A, Gormally S, Saxena R. Copper-associated liver disease in childhood. J Hepatol 1995;25:538-543. [ Links ]
11. Hurslen SP, Tanner MS, Lyon TD, Fell GS, Lowry MF. Copper associated childhood cirrhosis. Gut 1994;35:1497-1500. [ Links ]
12. Maggiore G, DeGiacomo C, Sessa F, Burgio GR. Idiopathic hepatic copper toxicosis in Chile. J Pediatr Gastroenteral Nutr 1987;6:980-983. [ Links ]
13. Pandit AN, Bhave S. Present interpretation of the role of copper in Indian childhood cirrhosis. Am J Clin Nutr 1996;63:830-835. [ Links ]
14. Walker SJ, Blomfield J. Wilson's disease or chronic copper poisoning. Arch Dis Child 1973;48:476-479. [ Links ]

