










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versão impressa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.66 no.6 México Nov./Dez. 2009
Caso clínico
Síndrome de hiperinmunoglobulinemia E. Reporte de dos casos
Hyperimmunoglobulin E syndrome: report of two cases
Miriam Puebla-Miranda1, Eduwiges Martínez-Luna2, María Elisa Vega-Memije2
1 Departamento de Dermatología Hospital General "Dr. Manuel Gea González", Secretaría de Salud, México, D. R, México.
2 Servicio de Dermatopatología, Hospital General "Dr. Manuel Gea González", Secretaría de Salud, México, D. R, México.
Autor de correspondencia:
María Elisa Vega Memije
Correo electrónico: dra_elisa_vega@yahoo.com.mx
Fecha de recepción: 29-04-08
Fecha de aprobación: 29-01-09
Resumen
Introducción. El síndrome de hiperinmunoglobulinemia E es una inmunodeficiencia sistémica poco frecuente, caracterizada por dermatitis eccematosa, abscesos fríos recurrentes, infecciones pulmonares con formación de neumatoceles, facies tosca, niveles elevados de inmunoglobulina E (IgE) en suero y eosinofilia.
Casos clínicos. Caso 1. Femenino de 11 años de edad con antecedentes de neumonía recurrente, gastroenteritis de repetición, dermatitis eccematosa de predominio en pliegues, y abscesos fríos; en estudios de laboratorio destacó el hallazgo de 16 070 eosinófilos e IgE de 4 864 Ul. Manejada con gammaglobulina se observó buena respuesta clínica. Caso 2. Femenino de 12 años de edad con historia de otitis recurrente y conjuntivitis supurativa, presentaba eccema crónico generalizado e impetiginizado. En estudios de laboratorio se reportó IgE de 3 000 UI; fue manejada con dapsona, trimetropim/sulfametoxazol y metotrexate.
Conclusión. Los 2 casos aquí informados presentaron piel eccematosa, infecciones recurrentes e incremento de los niveles de IgE, compatibles con síndrome de hiperinmunoglobulinemia E en la forma autosómica recesiva.
Palabras clave: Síndrome de hiperinmunoglobulinemia E, síndrome de Job, eccema, infección pulmonar, abscesos fríos, inmunodeficiencia congénita.
Abstract
Background. Hyperimmunoglobulin E syndrome is a rare systemic immunodeficiency characterized by eczematous dermatitis, recurrent cold abscesses, lung infections with pneumatoceles, coarse facial appearance, high IgE levels and eosinophilia.
Case reports. Case 1: We report the case of an 11-year-old female with a history of recurrent lung infections, recurrent gastroenteritis, eczematous dermatitis affecting the skin folds and cold abscesses. Laboratory studies showed elevated eosinophils (16 070) and IgE 4864 IU. The patient received treatment with g-globulin, showing adequate clinic response to treatment. Case 2: We present the case of a 12-year-old female with a history of recurrent otitis and suppurative conjunctivitis, showing widespread and chronic infected eczema. Laboratory studies showed elevated IgE (3 000 IU). She was treated with dapsone, trimethoprim/sulfamethoxazole and methotrexate.
Conclusions. We presented two patients with eczematous skin, recurrent infections and increased IgE levels, which are compatible with hyperimmunoglobulin E autosomal recessive syndrome.
Key words: hyperimmunoglobulin E syndrome, Job's syndrome, eczema, lung infection, cold abscesses, congenital immunodeficiency.
Introducción
El síndrome de hiperinmunoglobulinemia E fue descrito por primera vez en 1966 por Davis y col.1 al reportar dos niñas con pelo rojo, dermatitis crónica severa, abscesos fríos y neumonía recurrente, denominando a esta enfermedad síndrome de Job en alusión al personaje bíblico Job, cuya fe se probó al soportar úlceras y fístulas que drenaron de por vida. Posteriormente, en 1972, Buckley y col.2 describieron este síndrome en dos niños con dermatitis severa, abscesos recurrentes a nivel cutáneo, pulmonar y articular, retardo en el crecimiento, facies tosca e hipersensibilidad inmediata exagerada asociada con niveles elevados de inmunoglobulina E (IgE) en suero y eosinofilia, por lo que también se le conoce como síndrome de Buckley; sin embargo, recientemente se trata de omitir los epónimos y se recomienda llamarlo síndrome de hiperinmunoglobulinemia E o síndrome hiper IgE. Esta alteración es producida por un defecto genético que ocasiona la producción de niveles séricos elevados de anticuerpos IgE; el eccema e infecciones recurrentes son característicos. Es una enfermedad extremadamente rara, a la fecha solo existen aproximadamente 250 casos reportados en la literatura.3 En la mayoría de los casos se hereda de manera autosómica dominante y predominan las manifestaciones dentales y alteraciones óseas;4 la forma de herencia autosómica recesiva se asocia a infecciones fúngicas o virales graves. El uso de gammaglobulina intravenosa a altas dosis ha sido eficaz en el tratamiento de esta enfermedad. En este artículo se informa de dos casos de síndrome de hiperinmunoglobulinemia E en su forma autosómica recesiva.56
Presentación de los casos clínicos
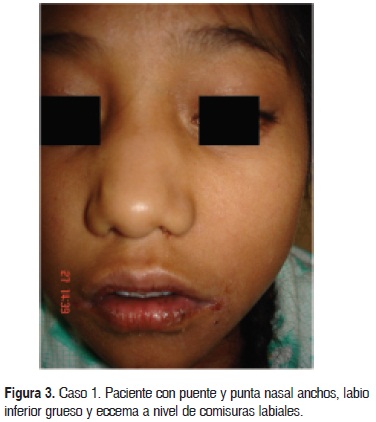
Caso 1. Paciente del sexo femenino de 11 años de edad, originaria y residente de Zimapam, Hidalgo, que se encontraba hospitalizada en una institución de tercer nivel con diagnóstico de neumonía (Fig. 1). Se solicitó interconsulta al Servicio de Dermatología por presentar dermatosis diseminada bilateral y simétrica, que afectaba las comisuras de la boca, cuello, pliegues axilares, antecubitales, inguinales y vulva; la dermatosis estaba constituida por grandes placas mal definidas, conformadas por eritema, costras melicéricas y hemáticas, exulceración y piel de aspecto eccematoso; en otras áreas predominaba la liquenificación, las costras hemáticas y la hiperpigmentación residual. A nivel axilar e inguinal había aumento de volumen y fístulas con salida de material purulento sin aumento de temperatura o eritema local (abscesos fríos) (Fig. 2). Presentaba desnutrición con peso de 23 kg y talla de 125 cm; a nivel de cara con puente nasal ancho y punta redonda (Fig. 3).

Al interrogatorio indirecto se refirió exantema al nacimiento, el cual mejoró con tratamiento tópico no especificado y desde los cinco años de edad presentó lesiones similares a las ya descritas, cursando con remisiones parciales y exacerbaciones.
En los resultados de laboratorio destacó la eosinofilia de 82% (0-7% normal) con una cuenta absoluta de 16 070 (0-0.8/μL) eosinófilos, y la inmunoglobulina E de 4 864 UI/mL (0-100 UI/mL); la interleucina (I L)-12 y la IL-2 estaban disminuidas y el interferón gamma y la IL-4 se reportaron levemente aumentados, con valores de células NK normales. El coproparasitoscópico reveló quistes de Giardia lamblia, para lo cual se dio tratamiento con metronidazol.
Con estos datos se consideró inidalmente el diagnóstico de eccema atópico; sin embargo, llamaba la atención el antecedente de dos hospitalizaciones a los cuatro y siete años de edad por neumonía, manejada con claritromicina, ketoconazol y cefatoxirma. Además de tener el antecedente de gastroenteritis de repetición desde los cinco años de edad con evacuaciones fétidas y con moco. A los 10 años de edad presentó abscesos fríos en piel y en el año 2005 se le había realizado cultivo de un ganglio inguinal que reportó adiaspiromicosis PAS positiva por Chysosporium parvum. En el año 2006 se le realizó biopsia pulmonar por una imagen radiopaca en radiografía de tórax, reportando lesión granulomatosa. La intradermorreacción con PPD fue negativa.
Al interrogatorio dirigido se negó consanguinidad de los padres, siendo producto de la gesta VI11, con dos hermanos finados, uno al año de edad por padecimiento dermatológico no especificado y el otro por cuadro séptico; el resto de los hermanos los refirieron como sanos.
Con lo anterior se realizó una biopsia cutánea que sugirió los diagnósticos de eccema secundario a síndrome de hiperinmunoglobulinemia E contra enfermedad granulomatosa crónica.
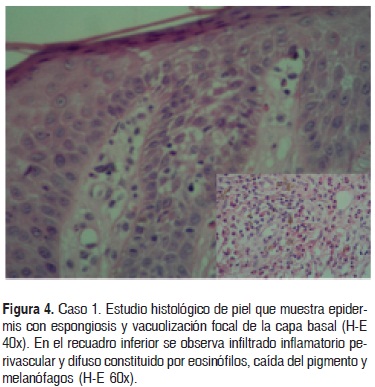
La histopatología de piel mostró espongiosis, vacuolización focal de la capa basal, exodtosis de eosinófilos, infiltrado inflamatorio superficial y profundo, perivascular e intersticial, constituido por eosinófilos, algunos linfocitos e histiocitos (Fig. 4).
Se integró el diagnóstico de síndrome de hiperinmunoglobulinemia E. La paciente fue tratada durante su hospitalización con ceftriaxona y amikacina por la neumonía, y a nivel tópico para el eccema, con mupirocina mezclado con aceponato de metilprednisolona dos veces por día y con fomentos secantes, observando buena respuesta al tratamiento. Se egresó con profilaxis a base de trimetoprim/sulfametoxazol e itraconazol y se inició gammaglobulina a dosis de 400 mg/kg de peso.
Posteriormente, a los tres meses de su egreso, la paciente fue hospitalizada por bronquiectasias infectadas; el cultivo de expectoración reportó Entamoeba coli y Branhamella catarrhalis. Presentó eosinofilia de 90% con 17 480 eosinófilos totales; recibió tratamiento con ceftriaxona, amikacina, gammaglobulina a 500 mg/kg de peso, y a su egreso se decidió aumentar la gammaglobulina a 1 g/kg de peso, con buena respuesta clínica.
Caso 2. Paciente femenino de 12 años de edad originaria y residente de Oaxaca, Oaxaca, hospitalizada en pediatría con los diagnósticos de impetigo generalizado, conjuntivitis y otitis media supurativa.
Se interconsultó al servicio de dermatología por presentar dermatosis generalizada, bilateral y simétrica, afectando todos los segmentos corporales, constituida en piel cabelluda por placas pseudoalopécicas, escama, costras serosanguíneas y melicéricas, en el resto se observaba con pápulas, pústulas y placas eccematosas impetiginizadas, así como maceración de pliegues (Fig. 5). A la exploración física presentaba adenopatías submaxilares, axilares e inguinales, con cinco meses de evolución, iniciando la dermatosis en pliegues, con diseminación progresiva; fue manejada con fomentos de sulfato de cobre 1:1000, didoxadlina, cloramfenicol oftálmico y loratadina/betametasona a 5 mg/0.25mg cada ocho horas. En los estudios de laboratorio resaltaba: leucocitosis, eosinofilia y bandemia; con cultivo de exudado positivo para E. coli. Se decidió su egreso por mejoría, con impresión diagnóstica de dermatitis atópica impetiginizada.
Dos meses después acudió nuevamente con fiebre no cuantificada, hiperemia conjuntival, secreción ocular verdosa, otitis externa y eccema generalizado, por lo que nuevamente se hospitalizó. Fue manejada con fomentos de sulfato de cobre, vioformoydidoxacilina por la sospecha diagnóstica de dermatitis atópica impetiginizada (cultivo positivo para Staphyloccocus aureus y E. coli); se agregó hidroxicina 1 mg/kg/día, prednisona 0.7 mg/kg/día e isoniacida a 300 mg/día como profilaxis para tuberculosis por el antecedente de Combe +. Se realizó la toma de biopsia para estudio histopatológico, en donde destacó la espongiosis de la epidermis e infiltrado inflamatorio por linfocitos y eosinófilos.
Ante los antecedentes de otitis externa recurrente (Fig. 6), motivo por el cual la paciente acudía regularmente al Servicio de Otorrinolaringología, de los episodios de eccema generalizado impetiginizado, así como de conjuntivitis, se estableció el diagnóstico probable de síndrome de hiperinmunoglobulinemia E, y se determinaron niveles de IgE séricos con un valor de 3 000 UI (0-100 UI/mL valor de referencia), con lo que se confirmó el diagnóstico. Fue egresada con manejo de dapsona 100 mg/ día, trimetropim/sulfametoxazol 160/800 mg/día y metotrexate; la paciente no acudió posteriormente para continuar seguimiento. No se registró la existencia de consanguinidad de los padres u otros antecedentes familiares de importancia.

Discusión
El síndrome de hiperinmunoglobulinemia E es una inmunodeficiencia sistémica rara, caracterizada por la tríada clásica de niveles altos de inmunoglobulina E, eccema e infecciones recurrentes. Presenta una incidencia de 1 en 500 000 nacidos vivos, sin predominio de género; hasta la fecha existen aproximadamente 250 casos reportados en la literatura.3 La forma de transmisión más frecuente es la autosómica dominante con penetrando variable. Por estudios citogenéticos se ha sugerido que la alteración se encuentra a nivel del brazo largo del cromosoma 4, predominando las manifestaciones dentales y alteraciones óseas.4 En cuanto a la transmisión autosómica recesiva, se ha puntualizado la mutación del gen Tyk2, caracterizándose por presentar infecciones fúngicas o virales graves y manifestaciones del sistema nervioso centraren donde puede ocurrir infarto isquémico, hemorragia subaracnoidea y hemiplejía.56 En estudios recientes se han precisado como determinantes en la etiología de este síndrome, en su forma dominante o esporádica, mutaciones en el gen STAT3; este gen interviene directamente en la respuesta de los monocitos a la IL-6, en consecuencia se produce disminución de la proteína 1 quimioatrayente de monocitos, además juega un papel en el desarrollo y diferenciación de células B y Th 17, y en la señalización de otras atocinas como la IL-10e IL-17.7,11
En estos pacientes existe disminución en la quimiotaxis de neutrófilos. Se considera que en su patogénesis interviene la disminución de receptores C3b en neutrófilos, así como la disminución de la molécula de adhesión L-selectina en los granulocitosylinfocitos.12
La elevación de IgE en suero se asocia específicamente con la formación de anticuerpos contra S. aureus; es característico encontrar niveles de IgE de más de 20 veces su valor normal (> 2000 UI/mL), con una cuenta elevada de eosinófilos en sangre. La eosinofilia se asocia con un incremento en la producción de GM-CSF y disminución deTGF-β. La IL-4 en general es normal. Además hay disminución de la producción de interferón y y factor de necrosis tumoral, con una pobre respuesta a la estimulación por IL-12, disminución en la población de linfocitos B de memoria y de la respuesta de hipersensibilidad tardía. Existe un aumento en la producción de intermediarios reactivos de oxígeno por los neutrófilos, lo que explica el daño tisular.13,14
En los estudios realizados se han descrito las alteraciones asociadas al síndrome, con la tríada clásica de abscesos de la piel y neumonías recurrentes, además de niveles elevados de IgE; el eccema severo es un hecho universal.15,16 Así mismo, se describe una facies tosca característica, sin que la apariencia sea completamente dismórfica; las características eraneofacíales más relevantes son las cejas prominentes, con la apariencia de ojos hundidos, puente y punta nasal anchos, labio inferior grueso, asimetría facial, frente prominente, hipertelorismo, prognatismo y piel facial áspera con poros dilatados.17 Sólo la paciente del primer caso presentaba estas características.
En un intento por establecer datos tempranos de la enfermedad se ha observado que 74% de los pacientes presentan prurito antes de los dos años de vida, 72% tiene historia de eccema flexural, y 81 % antecedente de exantema del recién nacido durante el primer mes de vida. Aunque aún no están establecidos los datos tempranos para el diagnóstico debido a que inicialmente no se presentan las principales características, como la dermatitis eccematosa, algunos autores proponen tomar en cuenta, dentro de los diagnósticos diferenciales de exantemas máculovésiculo-pustulares en neonatos, al síndrome de hiperinmunoglobulinemia E.18,20 La paciente del caso uno tiene el antecedente de exantema al nacimiento y, al momento de la interconsulta de eccema y liquenificación, lesiones frecuentes en pacientes con dermatitis atópica; en la paciente del caso dos, además del eccema con impetiginización secundaria, sobresale el antecedente de infecciones óticas recurrentes y conjuntivitis. Los pacientes con síndrome de hiperinmunoglobulinemia E autosómica recesiva presentan con más frecuencia infecciones virales por molusco contagioso y herpes simple; además, tienen una alta frecuencia de alteraciones del sistema nervioso central.5 Estos datos no se encontraron en los casos que aquí se informan.
Entre las alteraciones dentales reportadas están la retención primaria de dientes, o no erupción de dientes permanentes, la presencia de dientes primarios y dientes permanentes al mismo tiempo y elevación del paladar; la paciente del primer caso tuvo historia de retardo en la dentición; sin embargo, a la exploración física no se encontraron alteraciones dentales.21,22
Con respecto al tratamiento, no hay uno curativo aún, por lo que las decisiones terapéuticas se basan en las manifestaciones clínicas. La terapéutica más efectiva es el uso de antibióticos sistémicos profilácticos, con cobertura dirigida contra S. aureus. Hay reportes de casos de mejoría clínica e inmunológica de la dermatitis eccematosa, disminución en la frecuencia de abscesos fríos, de IgE y de la eosinofilia con el uso continuo detrimetoprim/sulfametoxazol (40 mg/kg/día/8 mg/kg/día); sin embargo, los efectos evaluados in vitro de este antibiótico a nivel de la quimiotaxis de neutrófilos y producción de hidrógeno, no fueron reproducibles y se desconoce hasta el momento el mecanismo de acción.23,24 Se ha reportado el uso de inmunoglobulina intravenosa en algunos casos, con resultados poco satisfactorios;25sin embargo, otro estudio realizado por Kirmata26 menciona una adecuada respuesta al manejo con gammaglobulina intravenosa a altas dosis (400 mg/kg/día) durante 5 días, se observa reducción en la severidad del eccema, además de presentar disminución de la producción de IgE in vivo e in vitro.27 ,29 Otro fármaco utilizado es el interferón gamma, que incrementa el índice quimiotáctico basal.27 Existen reportes de casos con éxito terapéutico secundario al uso de isotretinoina, sin que hasta el momento se conozca el posible mecanismo de acción.28
Respecto a la predisposición a malignidad, hasta la fecha no se ha probado que ésta aumente; sólo se han reportado casos que mencionan alguna asociación con neoplasias hematológicas.30,34
Referencias
1. Davis SD, Schaller J, Wedgwood RJ. Job's syndrome: recurrent, "cold", staphylococcal abscesses. Lancet 1966,1:1013-1015. [ Links ]
2. Buckley RH, Wray BB, Elmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics 1972,49:69-70. [ Links ]
3. De Witt CA, Bishop AB, Buescher L, Stone S. Hyperimmunoglobulin E syndrome: Two cases and review of the literature. J Am Acad Dermatol 2006,54:855-865. [ Links ]
4. Grimbacher B, Holland SM, Gallin Jl, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections. An autosomal dominant multisystem disorder. N Engl J Med 1999,340:692-702. [ Links ]
5. Renner ED, Puck J, Holland S, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome: A distinct disease entity. J Pediatr 2004; 144:93-99. [ Links ]
6. Verma S, Wollina U. Job's syndrome-a case report. JEADV 2003; 17:711-714. [ Links ]
7. MinegishiY, Karasuyama H. Genetic origins of hyper-IgE syndrome. Curr Allergy Asthma Rep 2008,8:386-391. [ Links ]
8. Holland S, DeLeo F, Elloumi H, Hsu A, Uzel G, Brodsky N, et al. STAT3 Mutations in the hyper-IgE syndrome. N Engl J Med 2007,357:1608-1619. [ Links ]
9. Paulson M, Freeman A, Holland S. Hyper IgE syndrome: an update on clinical aspects and the role of signal transducer and activator of transcription 3. Curr Opin Allergy Clin Immunol 2008,8:527-533. [ Links ]
10. Jiao H, Toth B, Erdos M, Fransson I, Rakoczi E, Balogh I, et al. Novel and recurrent STAT3 mutations in hyper-IgE syndrome patients from different ethnic groups. Mol Immunol 2008,46:202-206. [ Links ]
11. MilnerJD, BrenchleyJM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impared T(H)17 cell differentiation in subjects with autosoman dominant hyper-IgE syndrome. Nature 2008,452:773-776. [ Links ]
12. Vargas L, Rodríguez M, Forero C, Montoya F, Montoya C, Sorensen R, et al. Increase in granulocyte-macrophage-colony-stimulating factor secretion and the respiratory burst with decreased L-selectin expression in hyper-IgE syndrome patients. Ann Allergy Asthma Immunol 1999,83:245-251. [ Links ]
13. Stiehm E. Cytokine dysregulation in the hyperimmunoglobulinemia E syndrome. J Pediatr 2000,136:141-143. [ Links ]
14. Speckmann C, Enders A, Woellner C, Thiel D, Rensing-Ehl A, Schlesier M, et al. Reduced memory B cell in patient with hyper IgE syndrome. Clin Immunol 2008,129:448-454. [ Links ]
15. Freeman A, Holland S. The hyper-lgE syndromes. Immunol Allergy Clin North Am 2008,28:277-291. [ Links ]
16. Chamlin S, McCalmont T, Cunningham B, Esterly N, Lai C, Mallory S, et al. Cutaneous manifestations of hyper-IgE syndrome in infants and children. J Pediatr 2002,141:572-575. [ Links ]
17. Borges W, Hensley T, Carey J, Petrak B, Hill H. The face of Job. J Pediatr 1998,133:303-305. [ Links ]
18. Eberting CL, Davis J, Puck J, Holland S, Turner M. Dermatitis and the newborn rash of hyper-IgE syndrome. Arch Dermatol 2004; 140:11 19-125. [ Links ]
19. Kamei R, Honig R Neonatal Job's syndrome featuring a vesicular eruption. Pediatr Dermatol 1988,5:75-82. [ Links ]
20. Erlewin-Lajeunesse M. Hyperimmunoglobulin-E syndrome with recurrent infection: a review of current opinion and treatment. Pediatr Allergy Immunol 2000,11:133-141. [ Links ]
21. Freeman AF, Domingo DL, Holland SM. Hyper IgE (Job's) syndrome: a primary immune deficiency with oral manifestations. Oral Dis 2009; 15:2-7. [ Links ]
22. Aldous J, Olson G, Parkin M. Dental observations of hyper IgE disorder. J Clin Pediatr Dent 2007,32:69-72. [ Links ]
23. Hattori K, Hasui M, Masuda K, Masuda M, Ogino H, Kobayashi Y. Successful trimethoprimsulfamethoxazole therapy in a patient with hyperimmunoglobulin E syndrome. Acta Paediatr 1993,82:324-326. [ Links ]
24. Tanaka H, Ito R, Onodera N, Waga S. Efficacy of long-term sulfamethoxazoletrimethoprim therapy in a boy with hyperimmunoglobulin E syndrome. Tohoku J Exp Med 1998; 186:61-66. [ Links ]
25. Wakim M, Alazard M, Yajima A, Speight D, Saxon A, Stiehm R. High dose intravenous immunoglobin in atopic dermatitis and hyper-IgE syndrome. Ann Allergy Asthma Immunol 1998,81:153-158. [ Links ]
26. Kimata H. Highdose intravenous γ-globulin treatment for hyperimmunoglobulin E syndrome. J Allergy Clin Immunol 1995,95:771-774. [ Links ]
27. Jeppson J, Jaffe H, Hill H. Use of recombinant human inter-feron gamma to enhance neutrophil chemotactic responses in Job's syndrome of hyperimmunoglobulinemia E and recurrent infections. J Pediatr 1991,118:383-387. [ Links ]
28. Shuttleworth D, Holt P, Mathews N. Hyperimmunoglobulin E syndrome: Treatment with isotretinoin. Br J Dermatol 1988; 119:93-99. [ Links ]
29. Orozco C, Velazquez L, Méndez N, Augusto B, Solazar T Hyper IgE syndrome. Opportune diagnosis and management. Rev Alerg Mex 2008;55:38-45. [ Links ]
30. Lin SJ, Huang JL, Hsieh KH. Hodgkin's disease in a child with hyperimmunoglobulin E syndrome. Pediatr Hematol Oncol 1998; 15:451-454. [ Links ]
31. Bale J, Wilson J, Hill H. Fatal histiocytic lymphoma of the brain associated with hyperimmunoglobulin-E and recurrent infections. Cancer 1977,39:2386-2390. [ Links ]
32. Gorin L, Jeha S, Sullivan M. Burkitt's lymphoma developing in a 7 year-old boy with hyper-IgE syndrome. J Allergy Clin Immunol 1989;83:5-10. [ Links ]
33. Onal I, Kurt M, Altundag K, Aksoy S, Dincer M, Gullu I. Peripheral T-cell lymphoma and Job's syndrome: a rare association. Med Oncol 2006;23:141-144. [ Links ]
34. Amlot P, Green L Atopy and immunoglobulin E concentrations in Hodgkin's disease and other lymphomas. BMJ 1978; 1:327-329. [ Links ]

