










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.66 no.5 México sep./oct. 2009
Caso clínico patológico
Síndrome de Beckwith-Wiedemann
Beckwith-Wiedemann syndrome
Verónica Fabiola Morán-Barroso1, Constanza García-Delgado1, Mónica Villa-Guillen2, Eduardo Bracho-Blanchet3, Mario Perezpeña-Diazconti4
1 Departamento de Genética Médica
2 Departamento de Neonatología
3 Departamento de Cirugía
4 Departamento de Patología, Hospital Infantil de México Federico Gómez, México, D. F., México
Autor de correspondencia:
Dr. Mario Perezpeña Diazconti
Correo electrónico: mpdiazconti@gmail.com
Fecha de recepción: 10-02-2009.
Fecha de aprobación: 17-02-2009.
Resumen de la historia clínica
A'2007'22
Motivo de consulta: defecto de pared en la línea media. Antecedentes heredo familiares: madre de 25 años, sana, padre de 27 años, campesino, sano. Tres hermanas sanas, un primo hermano de cuatro años con atresia intestinal que se manejo quirúrgicamente. Antecedentes no patológicos: originario y residente del estado de Guerrero, de nivel socioeconómico bajo, en ayuno desde el nacimiento. Antecedentes perinatales: producto de gesta IV, con control prenatal irregular, sin administración de vitaminas ni ácido fólico; parto en domicilio atendido por partera a las 38 semanas de gestación, sin conocerse peso al nacimiento, talla ni valoración de Apgar. Se refirió a un hospital de segundo nivel de atención con peso de 3 250 g; se manejo con ayuno, soluciones intravenosas, antibióticos (ampicilina, amikacina), sonda nasogástrica abierta, trasladándose al Hospital Infantil de México Federico Gómez
Padecimiento final: defecto de pared desde el nacimiento.
Exploración física: se encontró activo, reactivo, mal hidratado. Tórax con tiros intercostales y disociación toracoabdominal, campos pulmonares con murmullo vesicular. En el corazón se auscultó soplo sistólico de eyección. En abdomen se observó defecto de pared en la línea media con onfalocele de 12 cm de diámetro y 6 cm de altura, con saco íntegro y vísceras en su interior. Se aplicó solución de isodine diluida en el saco y se colocaron gasas estériles.
Evolución: se realizó ecocardiograma que evidenció conducto arterioso pequeño y foramen oval permeable. Ingresó a terapia quirúrgica con cambio de ventilación mecánica a fase II, por dificultad respiratoria y desaturación, con FiO2 a 40%, PIM 17, PEEP 4, relación I:E 1:4, frecuencia respiratoria 40/min. Progresando rápidamente a modo SIM. El ultrasonido renal mostró hidronefrosis bilateral.
Exámenes de laboratorio: hemoglobina 19.7 g/dL, hematócrito 58%, leucocitos de 4 453/mm3, segmentados 4%, linfocitos 25%, bandas 7%, monocitos 20%, plaquetas de 165 000/mm3, VCM 103 fl, HCM 35 pg, urea 13 mg/dL, creatinina 0.5 mg/dL, Na 133 mEq/L, K 4.6 mEq/L, Cl 103 mEq/L, Ca 7.9 mg/dL, P 5.2 mg/dL, proteína C reactiva (PCR) 18.7 mg/dL; pH 7.31, PaO2 80.5, PaCO2 42.8, CO2T 17.8, HCO3 21.3, EB -3.8, SaO2 96.7%, lactato 1.1.
Presentó taquicardia, piel marmórea, datos de sobrecarga hídrica con tensión arterial sistólica entre 67-91 mm Hg; se agregó edema de miembros inferiores y se administró furosemida. Al practicar la curación del onfalocele se encontró fluido peritoneal en las gasas y secreción verde fétida, observándose retardo en el llenado capilar, secreción amarillenta por cánula traqueal, sangrado, a la auscultación estertores crepitantes, administrándose dobutamina y dos cargas rápidas sin respuesta. La radiografía de tórax mostró infiltrado basal derecho. En el cultivo de aspirado traqueal se aisló Pseudomonas aeruginosa. Presentó extrasístoles ventriculares, bradicardia y paro cardioventilatorio que no respondió a maniobras de reanimación.
Discusión del caso clínico
Dra. Mónica Villa Guillén (Unidad de Cuidados Intensivos Neonatales). El paciente que se discute el día de hoy cursó con onfalocele, el cual, en la en la mayoría de los casos es multifactorial, con una tasa de recurrencia de 1%; sin embargo, como defecto aislado se asocia más frecuentemente a un patrón hereditario que puede ser autosómico dominante, recesivo o ligado al cromosoma X, en cuyos casos la recurrencia es mayor. Llama la atención otro caso de malformación de tubo digestivo en la misma familia.
El paciente fue producto de la gesta IV con control prenatal irregular –sólo en tres ocasiones acudió a consulta–, sin ingesta de multivitamínicos ni ácido fólico. La Norma Oficial Mexicana establece desde el año 2003, que deben administrarse 400 µg diarios de ácido fólico a las mujeres en la etapa periconcepcional, esto es, de ser posible, tres meses antes del embarazo y hasta la semana 12 de gestación, con el fin de prevenir defectos congénitos. Actualmente está en curso la fortificación con ácido fólico de la tortilla, como parte del programa de la Red Nacional de Prevención de la Discapacidad. Está claramente establecida la asociación de la deficiencia de ácido fólico con defectos de tipo neural, como mielomengocele y anencefalia; no es tan claro, pero también se ha asociado a LPH, cardiopatías y defectos del tracto urinario. Mills y col.,1 en un estudio pequeño de 25 casos de onfalocele y 59 controles sin defectos congénitos, encontraron que el polimorfismo de un solo nucleótido de la enzima metiltetrahidrofolatoreductasa, que consiste en el cambio de citosina por timina en la posición 677, incrementó tres veces el riesgo de presentar onfalocele; esta variante reduce la actividad de la enzima, sugiriendo un mecanismo por el cual el ácido fólico puede prevenir la presentación de este problema. Botto y col.2 reportaron que el uso periconcepcional de multivitamínicos disminuyó en 60% el riesgo de onfalocele no sindromático en 72 casos de onfalocele y 3 029 controles de nacimientos en el área metropolitana de Atlanta entre 1968 y 1980. Regresando a nuestro caso, en el sexto mes de embarazo se hizo el diagnóstico por ultrasonido de defecto de pared abdominal. Éste es el estándar de oro para el diagnóstico prenatal, pero la tasa de detección varía de 60 a 90%, lo cual depende del equipo utilizado, pero sobre todo del operador. Es importante enfatizar que el diagnóstico sólo puede conformarse después de la semana 12-13 de gestación, ya que antes se considera fisiológica la herniación del intestino (la hernia-ción fisiológica no mide más de 7 mm de diámetro y no debe ser aparente en fetos con longitud crown-rump >44 cm).
El ultrasonido también es útil para detectar anomalías asociadas, las cuales pueden ser múltiples y se presentan en más de 70% de los casos. Las anomalías cromosómicas se presentan en 40-60% de los casos y pueden ser: trisomías 13, 8, y 21, síndrome de Turner, de Klinefelter, o de triploidia. Idealmente, a todo paciente en quien se detecta por ultrasonido un onfalocele, debe de realizársele una amniocentesis.
Otras alteraciones que pueden presentarse son: cardiacas (16-47%), genitourinarias (40%), musculoesqueléticas (10-30%) y neurológicas; o presentarse con síndromes asociados como el de Beckwith-Wiedemann (5-10%), onfalocele, macroglosia, visceromegalias, hipoglucemia.
Estos casos deben referirse y manejarse desde el nacimiento en un centro de tercer nivel; sin embargo, este paciente, a pesar del diagnóstico prenatal, nació en su domicilio por vía vaginal. En general no hay evidencia suficiente que apoye el nacimiento por cesárea, salvo en los onfaloceles gigantes como era el caso, en donde con un defecto tan grande puede complicarse con un parto distócico y asfixia perinatal. No hay datos en la historia clínica para apoyar alguna de estas situaciones; otro evento adverso para el paciente fueron los tres traslados en 48 horas, primero al centro de salud, después al Hospital General de Chilpancingo y, finalmente, a nuestro hospital. Sabemos que fue manejado con ayuno, antibióticos, sin especificarse cómo se manejó el defecto abdominal. Es importante este aspecto, porque aunque el onfalocele no estaba expuesto al 100%, ya que tenía un saco que lo protegía, era importante cubrirlo. Ya hay disponibles, aunque no en nuestro medio, bolsas estériles especiales para gastrosquisis y onfalocele que disminuyen las pérdidas insensibles, las cuales en estos casos están muy aumentadas; por otro lado, previene las infecciones y permite visualizar continuamente el defecto. Otra opción es la protección con gasas húmedas estériles, aunque esta técnica deja residuos, absorbe líquido y puede lesionar el defecto.
Cuando llegó a esta institución se corroboró el diagnóstico de onfalocele gigante, que se define así cuando el diámetro es mayor de 5 cm; este defecto medía 12 cm con saco íntegro y vísceras en su interior, sin especificar cuáles, aunque estos defectos tan grandes habitualmente contienen, además de intestino, hígado y otros órganos.
De la exploración al ingreso, llama la atención la dificultad respiratoria, la presencia de soplo, taquicardia y deshidratación isonatrémica. La dificultad respiratoria pudo haber sido de origen infeccioso, con o sin foco pulmonar, metabólico (presencia de hipocalcemia), restrictivo por el onfalocele gigante, o secundario a hipoplasia pulmonar; anormalidades que se han asociado a este defecto de pared abdominal y que son datos de mal pronóstico. La radiografía se describe como normal, por lo que se descarta neumonía e hipoplasia en ese momento de la evolución. La hemoglobina de 18 g/dL es normal para el segundo día de vida, aunque está en límites superiores, lo que puede explicarse por la deshidratación; tenía leucocitos bajos para su edad (mínimo 9 000/mm3), bandemia de 50%, relación I/T 0.7, monocitosis, plaquetas en límites inferiores normales, hipocalcemia e hipoalbuminemia. La gasometría mostró acidosis metabólica leve con hiperlactatemia. En ese momento, además de la deshidratación, el paciente tenía datos de infección, que pudo ser temprana o nosocomial de adquisición en el hospital de referencia, pues ya contaba con 48 horas de vida. El paciente ameritaba cambio de esquema antibiótico y determinación de plaquetas como parte importante del protocolo de sepsis en el recién nacido.
En cuanto a la deshidratación, se asumió ésta como de 10%, isonatrémica por el manejo que se dio con soluciones (1:1), con un cálculo de déficit de solutos de 28 mOsm/kg, de los cuales la mitad correspondían a sodio. La deshidratación era leve, no hubo pérdida de peso, sólo se reportó edema de una de las extremidades; la acidosis fue leve, la urea estaba ligeramente elevada (16, con valores normales de 13 en la primera semana de vida), y pudo haberse manejado con soluciones a 150 mL/kg (2:1), con aporte de sodio a 7 mEq/L (requerimientos basales de 80 mL/kg más 50% extra de pérdidas insensibles, 10 mL/kg =90 mL/ kg + 50 mL/kg por deshidratación).
Llama la atención la hipoalbuminemia de 1.5 g, situación frecuente en estos pacientes, y a la cual debe darse seguimiento, ya que valores menores son indicación de administración de albúmina con el fin de evitar la fuga de líquido al tercer espacio a todos niveles; el día de su muerte ya tenía 0.6 g, aunque no contamos con valores de días previos.
También quiero hacer mención especial a la monocitosis que durante toda su estancia presentó este paciente. Es importante recordar las causas de la misma, y si bien, la inflamación a nivel abdominal puede explicar este hallazgo, debemos tener presentes entidades como: sífilis (no se realizó VDRL), infección por Listeria monocytogenes, infecciones respiratorias por microorganismos atípicos como Chlamydia y Micoplasma; y en general procesos infecciosos e inflamatorios crónicos.
El ecocardiograma reveló la presencia de conducto arterioso, y el electrocardiograma fue normal salvo por el QTc que estaba alargado en 0.36, cuando no debería ser mayor a 0.24; este hallazgo se explica probablemente por la hipocalcemia.
De otros estudios de extensión, el ultrasonido transfontanelar mostró la presencia de ventrículos colapsados; este hallazgo se ve en pacientes con edema cerebral, pudiendo haber estado condicionado en este caso por asfixia perinatal o por la hipoalbuminemia con la que cursaba. El ultrasonido renal mostró morfología y dimensiones normales con hidronefrosis bilateral; dentro de las anomalías genitourinarias reportadas asociadas a onfalocele está la obstrucción de la unión urete-ropiélica, que pudo ser la causa de la hidronefro-sis, o bien, que el aumento de la presión intraabdominal (con la que cursa esta patología) haya comprimido los riñones y provocado obstrucción al flujo urinario.
El paciente evolucionó a la gravedad, al tercer día de vida y primero de hospitalización requirió de intubación por dificultad respiratoria y acidosis respiratoria; en la biometría hemática, los leucocitos estaban en menos de 5 000/mm3, y la PCR en 18.7. En la literatura hay más de 160 referencias de PCR en neonatos, y sólo dos son estudios aleatorizados controlados. A pesar de esta vasta literatura, a la fecha no se ha establecido el estándar de práctica para el uso de PCR en la evaluación de sepsis neonatal. La PCR puede aumentar en varias situaciones clínicas no infecciosas; así, Ishibachi y col., encontraron que de 24 a 48 horas después del parto vaginal, los niveles pueden exceder los 10 mg/L. Debido a que la PCR es reactante o molécula de fase aguda inespecífica, numerosas causas de inflamación y daño tisular pueden incrementarla, como en el presente caso. En un estudio retrospectivo, realizado por Benitz y col.3 en la Universidad de Stanford, las mediciones seriadas de PCR tuvieron un valor predictivo negativo de 99% para sepsis. Dos concentraciones de PCR menores a 10 mg/dL obtenidas cada 24 horas, 8 a 48 horas después de la presentación del cuadro, hicieron la posibilidad de sepsis muy improbable.
Desafortunadamente, no hay un marcador único y definitivo de sepsis neonatal, y se debe realizar una evaluación de todos los elementos con los que contamos para establecer el diagnóstico. El paciente no evolucionó bien, ni por los datos clínicos ni por los resultados de laboratorio. Para el día 30 de marzo, ya con dos días de hospitalización, persistió con acidosis respiratoria, con leucopenia y con disminución en las plaquetas; el día primero de abril estaba taquicárdico, con piel marmórea, y se indicó cefotaxime; para el día dos requirió de incremento en los parámetros de ventilación, y por sospecha de neumonía se indicó dicloxacilina; el día tres presentó datos de peritonitis y estado de choque; hasta este momento se agregó carbapenem. Posiblemente desde el día 30 de marzo, con 48 horas de hospitalización, ameritaba manejo antimicrobiano más agresivo, ya que la infección no estaba controlada y lo más probable es que se había agregado otro proceso de adquisición en nuestro hospital. Queda la duda si la peritonitis no se detectó a tiempo, y con la cobertura inadecuada fue lo que al final determinó la muerte de este paciente.
Las infecciones nosocomiales son un problema grave en las salas de terapia intensiva neonatal. El empleo de catéteres, alimentación parenteral, ventiladores, y antibióticos de amplio espectro han dado lugar a un fenómeno propicio para la invasión bacteriana, y los recién nacidos son huéspedes especialmente vulnerables. Del año 2002 al 2006, la tasa de infecciones nosocomiales fue de 49/100 egresos en la Unidad de Cuidados Intensivos Neonatales (UCIN) (uno de cada dos pacientes tiene una infección nosocomial). Si consideramos la epidemiología de este hospital, los patrones de resistencia y al huésped, debemos cubrir los gérmenes gram negativos, que representan 70% de las infecciones nosocomiales en la Terapia Quirúrgica, con Klebsiella y Pseudomonas a la cabeza; los gram positivos con 20%, la mayoría ECON, con 96% de meticilino-resistencia; y el resto Candida, en su mayoría albicans. Un huésped con defecto de pared abdominal gravemente enfermo, requiere un esquema de imipenem-vancomicina para cubrir incluso gérmenes anaerobios asociados a infección peritoneal; la dicloxacilina no tiene cabida en el manejo de estos neonatos.
El día 4 de abril persistió el estado de choque, siendo la neumonía franca. Se reportó un cultivo de aspirado bronquial positivo para Pseudomonas, cuyo valor es muy cuestionable en el diagnóstico de neumonía asociada a ventilador debido a que pudo estar contaminado o reflejar simplemente colonización. De acuerdo a los criterios del CDC de octubre 2004, un cultivo de vías respiratorias debe ser cuantitativo, con valores mayores de 103 UFC/mL si se obtiene por broncoscopio, ya sea con lavado broncoalveolar, o con cepillado; también se aceptan estas técnicas de obtención de muestras sin broncoscopio. Además, igual o más de 5% de las células deben contener bacterias intracelulares en el examen microscópico directo (tinción de Gram). No quiero mencionar que Pseudomonas no pudo ser la causa de la infección, sólo dejo claro que el aspirado traqueal o bronquial no son métodos adecuados de diagnóstico.
El paciente requirió de volumen, aminas, con 30 000/mm3 de plaquetas, 30% de bandas, hiperlactatemia y problemas de coagulación. Finalmente, presentó extrasístoles ventriculares y bradicardia que no respondieron a maniobras de reanimación.
Los diagnósticos clínicos finales se incluyen en el cuadro 1.

Dr. Santos. El esquema antibiótico seleccionado inicialmente no era el adecuado, la presencia de monocitos es sugestivo más no diagnóstico de L. monocytogenes; sin embargo, en ningún momento se pensó en administrar ampicilina, que era lo que necesitaba ante esa consideración. En cambio, el día antes de que falleciera, se cambió a imepenen, y los niveles de monocitos seguían muy elevados, debiendo haberse cumplido el tratamiento para L. monocytogenes por la monocitosis.
Dr. Bracho. Dados los comentarios sobre los esquemas antibióticos, es pertinente la opinión del Dr. Rene Farfán, adscrito al Departamento de Infectología.
Dr. Farfán. El inicio de un esquema terapéutico empírico tiene que tomar en cuenta los factores de riesgo de cada uno de los pacientes. Si bien es cierto que siempre se ha dicho que ampicilina-amikacina es un esquema de elección de primera línea para el tratamiento de sepsis neonatal temprana, también se tienen que tomar en cuenta otros factores de riesgo, como: onfalocele, parto fortuito, ruptura prematura de membranas y traslado de otros hospitales. La monocitosis tiene un valor importante; un esquema probablemente adecuado al inicio de este paciente pudo haber sido una cefalosporina de tercera generación más la ampicilina para tratar de cubrir los gérmenes más habituales. Escalar un esquema de antibióticos depende mucho de la evolución clínica del paciente y de sus factores de riesgo.
Dr. Bracho. Le pedí al Dr. Anselmo Domínguez Chicas, adscrito al Departamento de Urología, un comentario acerca de la hidronefrosis postnatal no prenatal.
Dr. Domínguez. El estudio de hidronefrosis postnatal está en relación a los diagnósticos probables; nos interesan, sobre todo, las de tipo obstructivo que son las que van a requerir una cirugía con mayor posibilidad. Estamos hablando de obstrucción pielouretereal y uretero-vesical, de valvas de uretra posterior y de grado de reflujo vesicoureteral, que no es obstructivo pero puede requerir algún tratamiento.
De la hidronefrosis, básicamente nos interesa la dilatación, el diámetro anteroposterior de la pelvis renal, así como si hay dilatación de los cálices o adelgazamiento de la corteza renal, cualquiera de estos datos implica mayor gravedad. Si es unilateral se hace un ultrasonido para evaluar la hidronefrosis y determinar si hay dilatación de ureteros; si existe dilatación, hacemos un cistograma para descartar el reflujo, en caso de que no lo haya pensamos en obstrucción ureteropiélica, que es el diagnóstico más común. En este caso se solicita un gammagrama renal a las tres o cuatro semanas de vida para encontrar los hallazgos que nos den el diagnóstico, con base a una curva de eliminación retardada o una diferencia en la función renal relativa.
El caso de una obstrucción bilateral es una urgencia diagnóstica. Se realiza ultrasonido en cuanto el paciente lo permita, al primero o segundo día de vida; si hay dilatación de los ureteros se hace el cistograma para descartar el reflujo vesicoureteral. En caso de que no haya dilatación se piensa en obstrucción perilateral, realizándose un gammagrama renal en la segunda semana de vida para realizar el diagnóstico, y al final de esta segunda semana también determinamos el comportamiento de la creatinina sérica. Si la creatinina disminuye, la función renal está conservada y podemos hacer el gammagrama hasta la segunda o tercera semana; en caso de que la creatinina no disminuya, entonces tenemos que evaluar una nefrostomía bilateral para asegurar la función de los riñones. La decisión del momento quirúrgico la determinan los hallazgos. Si se confirma una obstrucción unilateral se puede operar después de los tres meses, si es bilateral conviene hacer por lo menos uno de los lados antes de los tres meses y, en particular, quiero mencionar que es importante detectar la hipertensión en la etapa neonatal; si se confirma, el procedimiento quirúrgico se tiene que hacer antes del mes de edad para garantizar el control de la hipertensión. Con respecto a este paciente, el hecho de que no hayan encontrado hidronefrosis en el ultrasonido prenatal sugiere que probablemente no era tan importante, y en la descripción del ultrasonido postnatal no se menciona cálices dilatados ni adelgazamiento de la corteza. Probablemente no era algo importante, tal vez una dilatación por pliegues fetales o un riñón fetal, que no implica patología alguna. Finalmente, la hidronefrosis que se sucede en los pacientes con gastrosquisis-onfalocele se presenta después de que se aplica el silo y se empieza a apretar, o después de que se cierra, que es cuando aumenta la presión intraabdominal; antes es poco frecuente.
Anatomía patológica
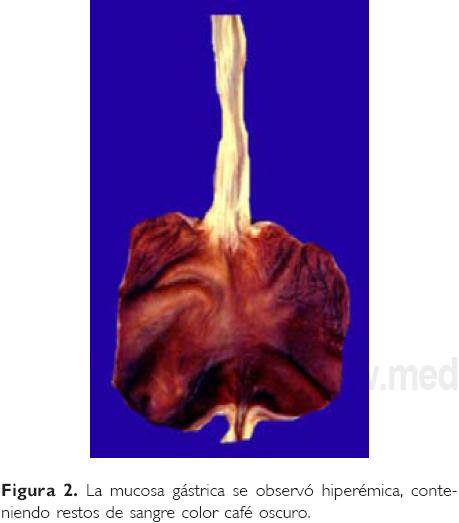
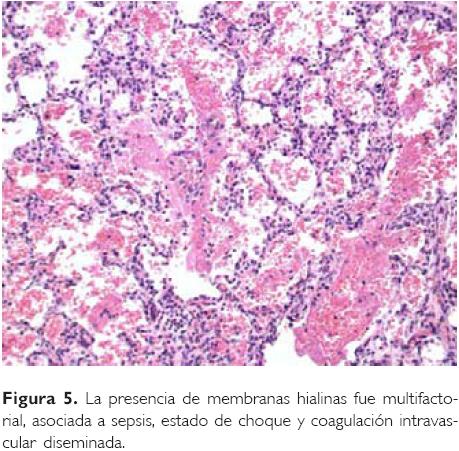
Dr. Mario Perezpeña Diazconti (Departamento de Patología). En el estudio post mortem, la facies del paciente presentaba macroglosia y surcos en los pabellones auriculares, neumotórax bilateral, en abdomen malrotación intestinal y el onfalocele, el cual midió 15 x 13 cm, con saco íntegro (Fig. 1), a través del cual se dibujaban las asas intestinales. Retirado el saco, éstas mostraban distensión, material purulento amarillo verdoso en la serosa, y la luz intestinal mostró la mucosa aplanada, de color rojo violáceo. En el estómago se encontraron restos de material hemático color café oscuro, y la mucosa era de color rojo vinoso (Fig. 2). Histológicamente, en la pared intestinal había tejido de granulación e infiltrado inflamatorio de leucocitos polimorfonucleares en la serosa. En el colon había fibrina e inflamación compuesta por linfocitos, células plasmáticas y células gigantes tipo cuerpo extraño. La mucosa estaba congestiva, con ulceración focal. En la pared abdominal había tejido fibroconectivo, necrosis coagulativa e intenso infiltrado inflamatorio de polimorfonucleares. Las glándulas suprarrenales mostraron citomegalia cortical (Fig. 3). El páncreas presentó hiperplasia de los islotes de Langerhans y de los acinos (Fig. 4). Los pulmones mostraron daño intenso con presencia de membranas hialinas (Fig. 5) y engrosamiento de la pared de arteriolas como dato de enfermedad vascular pulmonar (Fig. 6).




Con los hallazgos descritos se integra el diagnóstico de síndrome de Beckwith-Wiedemann. En el cuadro 2 se mencionan los diagnósticos anatómicos finales.

El síndrome de Beckwith-Wiedemann se caracteriza por macroglosia, hipertrofia y defecto de la pared abdominal, que incluye onfalocele, hernia umbilical o diastasis de recto.4 Los pacientes pueden estar afectados por hipoglucemia, por lo que la prevención puede evitar secuelas neurológicas graves. Hay macrosomía y polihidramnios.5,6 Las alteraciones faciales incluyen: nevo flameo, hipoplasia de la mitad de la cara y pabellones auriculares con indentaciones. Las alteraciones craneales son occipucio prominente y macroglosia. Hay malrotación intestinal, hepatomegalia, esplenomegalia y, ocasionalmente, cardiomegalia. La pancreatomegalia se presenta como hallazgo de autopsia. Se ha informado de hiperplasia uterina, ovárica, testicular, fálica, de vejiga, o clitoromegalia. La inteligencia es normal; cuando hay retardo en el desarrollo se asocia a duplicación cromosómica de 11p5, hipoglucemia no controlada o complicaciones de prematuridad.
Hasta 7.5% de los pacientes con Beckwith-Wiedemann desarrollan neoplasias,6,7 mucho más frecuentes en la región intraabdominal. Las más comunes son: tumor de Wilms, carcinoma de corteza suprarrenal y hepatoblastoma. El rabdomiosarcoma y neuroblastoma muestran aumento de la frecuencia. El incremento de la frecuencia de tumor de Wilms en el síndrome de Beckwith-Wiedemann está asociado a la hemi-hipertrofia, nefromegalia, restos nefrogénicos o nefroblastomatosis y a los subgrupos con disomia uniparental 11p5 e hipermetilación de H19.
La hipoglucemia se presenta hasta en 63%, es transitoria y responde a terapia. Ocurre regresión espontánea alrededor de los cuatro años. Puede haber policitemia hasta en 20% de los casos, hipocalcemia, hipercolesterolemia e hiperlipidemia. La hipercalciuria se presenta hasta en 22% de los casos.
En este síndrome, la citomegalia se presenta en la corteza suprarrenal, con hiperplasia de la médula adrenal. En el páncreas hay hiperplasia de acinos, conductos e islotes de Langerhans. Puede haber hiperplasia de células de Leydig y adenomatosa de los ovarios.
Dr. Adrián Chávez (Unidad de Terapia Intensiva Pediátrica). El niño presentó síndrome de dificultad respiratoria aguda secundario a la sepsis abdominal y, con base en eso, el análisis crítico de nuestra actuación muestra que este niño debió ser ventilado con alta frecuencia desde que se intubó y no con ventilación convencional, puesto que también cursó con hipertensión arterial pulmonar.
Discusión de Genética Médica
Dra. García-Delgado. El propósitus tenía el antecedente de un primo con malformación de tubo digestivo, desconocemos si estaban relacionados por línea paterna o materna, pero este dato es relevante para analizar el cuadro clínico de estos niños, que si se diagnostican y se manejan a tiempo pueden sobrevivir. De 2 a 3% de los casos de síndrome de Beckwith-Wiedemann tienen anormalidades citogenéticas que involucran al cromosoma 11 en el brazo corto en la región 15, incluyendo duplicaciones derivadas del cromosoma paterno, inversiones y translocaciones maternas; sólo 15% tiene historia familiar y se presenta cuando se hereda por línea materna; 20% de los casos son esporádicos y hay una disomía uniparental del cromosoma 11, casi siempre asociados a hemi-hipertrofia. De 5 a 10% de los casos esporádicos tienen un patrón de mutilación anormal de los genes H19 e IGF2, es decir, que 80% de los casos son esporádicos y 20% o menos son familiares.5
Recordemos que en la disomía uniparental, un par de cromosomas homólogos son heredados de un solo progenitor; si hacemos estudio convencional de cromosomas, el número de estos es normal, sólo el estudio a nivel molecular detectará la alteración. El ejemplo clínico de esta entidad nosológica es la alteración en el cromosoma número 15, en donde, si la disomía es de origen materno, los pacientes presentan síndrome de Prader Willi (OMIM 176270), y si es de origen paterno tendrán síndrome de Angelman (OMIM 10583).6,7
En el caso del síndrome de Beckwith-Wiedemann, es importante recordar que se asocia a alteraciones de la impronta genómica, fenómeno en el cual la expresión del gen depende de cuál es su origen parental. La impronta genómica se conoce desde los años ochenta y se sabe que ciertas regiones del genoma no son funcionalmente equivalentes, dependiendo de su origen materno o paterno, y obviamente de modificaciones epigenéticas del genoma.8 En referencia a los genes relacionados con la presentación del síndrome de Beckwith-Wiedemann, con loci en el cromosoma número 11 en el brazo corto en la región 1, subregión 5, hay genes que tienen expresión diferencial, ya sea materna o paterna. Entre los genes principales de control de expresión de la región se considera a H19 (gen que codifica a un RNA no traducido) e IGF2 (gen que codifica al factor de crecimiento parecido a la insulina tipo 2), además del gen KVLQT1 (y del gen CDKN1C o LTI1, el cual codifica para un inhibidor de ciclinas dependiente de cinasa). Si está alterado, el gen H19 puede a su vez alterar la expresión de IGF2, ocasionando un sobrecrecimiento. Lo interesante es recordar que la "contraparte" del síndrome de Beckwith-Wiedemann es el síndrome de Silver Russel, en el que la hipometilación de H19, en casos de disomía uniparental materna, afectará de manera inversa la expresión del gen IGF2, y podría explicar el retardo en el crecimiento que se ve en el síndrome de Beckwith-Wiedemann; si bien, no es el único mecanismo molecular propuesto.9-15
En el caso de esta familia, lo que se hará para poder ofrecerles asesoramiento genético es realizar una historia clínica con un pedigree completo, cariotipo convencional a los padres y, dependiendo de esto, teóricamente podría hacerse estudio molecular del tejido de autopsia.
Referencias
1. O´Leary VB, Mills JL, Parle-McDermott A, Pangilinan F, Molloy AM, Cox C, et al. Screening for new MTHFR polymorphism and NTD risk. Am J Med Genet. 2005; 138A: 99-106. [ Links ]
2. Botto LD, Mulinare J, Erickson JD. Occurrence of omphalocele in relation to maternal multivitamin use: A population -based study. Pediatrics. 2002; 109: 904-8. [ Links ]
3. Benitz WE, Han MY, Madan A, Ramachandra P. Serial serum C-reactive levels in the diagnosis of neonatal infection. Pediatrics. 1998; 102: E41. [ Links ]
4. Sotelo-Ávila C, González-Crussi F, Fowler JW. Complete and incomplete forms of Beckwith-Wiedemann syndrome: Their oncogenic potential. J Pediatr. 1980; 96: 47. [ Links ]
5. Emery A, Rimoin DL. Principles and practice of medical genetics. En: Graham JM Jr., Rimoin DL, editores. Abnormal body size and proportion. 4ta ed. Vol. I. New York: Churchill Livingstone; 78-9. [ Links ]
6. Camprubi-Sánchez C. From the clinical to the genetic diagnosis of Prader-Willi and Angelman syndromes. Rev Neurol. 2006; 42: S61-7. [ Links ]
7. Kotzot D. Prenatal testing for uniparental disomy: Indications and clinical relevance. Ultrasound Obstet Gynecol. 2008; 31: 100-5. [ Links ]
8. Morison IM, Reeve AE. A catalogue of imprinted genes and parent-of-origin effects in humans and animals. Hum Mol Genet. 1998; 7: 1599-609. [ Links ]
9. Cohen MM Jr. Beckwith-Wiedemann syndrome: Historical, clinic-pathological, and etiopathogenetic perspectives. Pediatr Dev Pathol. 2005; 8: 287. [ Links ]
10. Beckwith JB. Children at increased risk for Wilms tumor: Monitoring issues. J Pediatr. 1998; 132: 377. [ Links ]
11. Wiedemann HR. Tumours and hemihypertrophy associated with Wiedemann-Beckwith syndrome. Eur J Pediatr. 1983; 141: 128. [ Links ]
12. www.ncbi.nlm.nih.gov/OMIM [ Links ]
13. Hall JG. Genomic imprinting: Review and relevance to human disease. Am J Hum Genet. 1990; 46: 857-73. [ Links ]
14.Cerrato F. The two-domain hypothesis in Beckwith-Wiedemann syndrome: Autonomous imprinting of the telomeric domain of the distal chromosome 7 cluster. Hum Mol Genet. 2005; 14: 503-11. [ Links ]
15. Cooper WNl. Molecular subtypes and phenotipic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2005; 13: 1025-32. [ Links ]

