










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.65 no.5 México sep./oct. 2008
Artículo original
Estudio clínico y genético del síndrome de Moebius
Clinic and genetic study of Moebius syndrome
Luis Gómez–Valencia, Anastasia Morales–Hernández, Ramón Miguel Cornelio–García, Ezequiel Toledo–Ocampo, María de los Remedios Briceño–González, Miriam Margot Rivera–Angles
Servicio de Genética, Hospital del Niño Dr. Rodolfo Nieto Padrón, Villahermosa, Tabasco; División Académica de Ciencias de la Salud, Universidad Juárez Autónoma de Tabasco, Villahermosa, Tabasco, México
Solicitud de sobretiros:
Dr. Luis Gómez Valencia
Servicio de Genética,
Hospital del Niño "Dr. Rodolfo Nieto Padrón",
Avenida Gregorio Méndez Magaña No. 2832,
Col. Tamulté, C. P. 86150, Villahermosa, Tabasco, México.
Fecha de recepción: 13–03–2008.
Fecha de aprobación: 30–07–2008.
Resumen
Introducción. Objetivo: analizar las manifestaciones clínicas fundamentales del síndrome de Moebius, su involucro genético y los factores que en nuestro medio se relacionan con esta entidad.
Métodos. En un estudio retrospectivo y descriptivo se analizaron los datos clínicos y de anamnesis familiar de 23 casos de niñas y niños con síndrome de Moebius, vistos en el Hospital del Niño de Villahermosa, Tabasco, en los últimos 20 años. Los criterios para el diagnóstico se establecieron con base a las manifestaciones clínicas, considerándose para ello a la inexpresividad facial, alteración de la succión y cierre palpebral incompleto con deficiente o nula visión lateral.
Resultados. La inexpresividad facial, la ptosis palpebral, pabellones auriculares grandes y la microglosia fueron los datos clínicos más representativos. En 2 pacientes se documentó la secuencia de Poland como entidad asociada. El análisis cromosómico realizado en 11 de los casos mostró normalidad cromosómica en el número y en la estructura. Todos los pacientes estudiados representaron casos esporádicos.
Conclusión. El diagnóstico temprano de la secuencia de Moebius favorece la planeación en el manejo del enfermo, y al mismo tiempo ofrece a los padres de los afectados un asesoramiento genético oportuno.
Palabras clave: Parálisis facial; síndrome de Moebius; inexpresividad facial.
Abstract
Introduction. Objective: to analyze the fundamental clinical manifestations of the Moebius syndrome, and the genetic factors related to this pathology.
Methods. In a retrospective descriptive study the clinical data and familiar anamnesis of 23 cases of girls and boys with Moebius syndrome attended in Children's Hospital of Villahermosa, Tabasco over the last 20 years, were analyzed. The criteria for the diagnosis were done on the basis of clinical manifestations, which included face inexpressiveness, alteration of the suction, and incomplete palpebral closure with partial or non–lateral vision.
Results. Face inexpressiveness, palpebral ptosis, big auricular pavilions and microglossia were the most representative clinical data. Two patients had Poland sequence in association to this pathology. Chromosomal analysis of the 11 cases showed normality in number and structure. All the studied patients represented sporadic cases.
Conclusion. An early diagnosis of Moebius syndrome favors an optimal patient management as well as an opportune genetic counseling.
Key words: Facial paralysis; hereditary diseases; Moebius syndrome; facial expression; face, inexpressiveness.
Introducción
El síndrome de Moebius es un trastorno no progresivo que se caracteriza por parálisis facial desde el nacimiento, debido a una agenesia o aplasia de los núcleos de los nervios craneales VI y VII, lo que provoca parálisis facial y estrabismo convergente.1 Villafranca y col.,2 señalan que esta malformación fue descrita inicialmente por von Graefe y Saemisch en 1880, Harlam en 1881 y Chrisholm en 1882, pero fue Moebius quien, en 1888, hizo un estudio completo de la enfermedad y en 1892 dio a conocer 43 casos de parálisis facial congénita y adquirida, de los cuales seis presentaban parálisis facial bilateral congénita y parálisis del VI par.2 En los últimos 110 años, el diagnóstico clínico se ha basado fundamentalmente en las distintas manifestaciones clínicas del trastorno y, para lo cual, se ha usado el término de síndrome de Moebius, refiriéndose a pacientes con involucro de varios pares de nervios craneales.3 La incidencia es de 1 por cada 10 000 nacimientos, afectando a hombres y mujeres por igual; y aunque la causa y la patogénesis exacta del síndrome sigue sin entenderse, se cree que representa a una rara displasia, debido a un proceso degenerativo.3,4 Los trabajos sobre genética, los hallazgos radiológicos y los datos de autopsia difieren en sus resultados acerca de las causas básicas del síndrome de Moebius.3 En el último siglo se han reportado más de 300 casos con esta entidad.3,4
El presente trabajo tiene como objetivo analizar las manifestaciones clínicas fundamentales del síndrome, su involucro genético y los factores que en nuestro medio se relacionan con esta enfermedad.
Métodos
En un estudio retrospectivo y descriptivo, se analizaron los expedientes de pacientes de ambos sexos con diagnóstico clínico de síndrome de Moebius que acudieron a la consulta del Servicio de Genética del Hospital del Niño "Dr. Rodolfo Nieto Padrón" de Villahermosa, Tabasco, México, durante los últimos 20 años, período comprendido de marzo de 1987 a febrero de 2007. Al total de los pacientes se les registró: lugar de procedencia, sexo, edad, peso al nacer, manifestaciones clínicas y complicaciones asociadas. Por parte de los padres se consignaron: edad, consanguinidad, número de gesta, semanas de gestación, exposición a teratógenos y tipo de parto. Para ubicar la procedencia de los pacientes se consideraron las características geográficas del estado de Tabasco. Los criterios para el diagnóstico se establecieron con base a las manifestaciones clínicas, considerándose para ello a la inexpresividad facial, alteración de la succión y cierre palpebral incompleto con deficiente o nula visión lateral. Todos los pacientes fueron valorados por los servicios de pediatría, neurología pediátrica, oftalmología y genética.
Resultados
Se estudiaron un total de 23 pacientes con diagnóstico de síndrome de Moebius; 15 fueron del género masculino y ocho del femenino. Trece pacientes acudieron al hospital para su valoración en los primeros seis meses de vida, nueve antes de los dos años de edad. Sólo un caso fue valorado por primera vez a la edad de siete años. Diez fueron productos de la primera gesta.
Con relación a la procedencia de los casos, todos los pacientes correspondieron al estado de Tabasco. En nueve, el peso al nacimiento estuvo comprendido entre los 2 500 y 3 500 g, y cinco presentaron peso por debajo de 2 500 g; 15 presentaron hipoxia al nacimiento. Al analizar las manifestaciones clínicas se encontró inexpresividad facial en todos los casos (Fig. 1), prominencia de pabellones auriculares en 19, microglosia en 15 y ptosis palpebral en 12 (Cuadro 1).

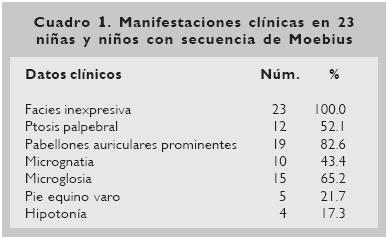
En dos casos se pudo documentar la secuencia de Poland como entidad asociada, así como labio hendido (1/23), malformación anorrectal (1/23), secuencia de Pierre–Robin (1/23) y comunicación interventricular (1/23). El análisis cromosómico se realizó en 11 pacientes, mostrando en todos normalidad cromosómica en el número y en la estructura de los mismos.
El 47.8% (11/23) de las mamás de estos niños y 65.2% (15/23) de los papás tenían edades entre los 20 y 30 años al momento del nacimiento de sus hijos. No se encontraron antecedentes de consanguinidad ni de padecimiento similar en otros miembros de la familia y se documentaron el antecedente de hipertensión materna (4/23), exposición a rayos "X" (1/23) y de misoprostol con carácter de abortivo (4/23); 56.5% de los nacimientos ocurrieron en medio hospitalario, atendidos por profesional médico y por parto eutócico.
Discusión
Stromland y col.,5 y Momtchilova y col.6 señalan que las anomalías dificultan la función de la expresión facial, la pronunciación, deglución y la succión. Cronemberger y col.,1 y Verzil y col.,7 por su parte, señalan que hay anomalías sistémicas como las malformaciones de miembros y el retardo mental, lo que hace sombrío el pronóstico de vida de estos niños.
Autores como Dooley y col.,8 y Miller y col.,9 en autopsias realizadas en niños con secuencia de Moebius, han documentado cuatro eventos de importancia: hipoplasia o ausencia de núcleos del mesencéfalo, degeneración destructiva de núcleos, que es lo que con mayor frecuencia se reporta, afección de nervios periféricos y presencia de alteraciones de fibras musculares de la región facial. Sin embargo, otros autores como Issaivanan y col.,10 al reportar el caso de una niña de un año de vida en donde coexistían Moebius–Poland–Klippel Feil, señalan la presencia de alteraciones morfológicas de la arteria subclavia, proponiendo que la ruptura temprana de la subclavia, alrededor de la sexta semana de desarrollo, pueden condicionar entidades como la que aquí se está presentando. Dentro de las entidades asociadas, se han reportado al síndrome de Klinefelter, el autismo infantil, secuencia de Poland y síndrome de Klipel Feil, entre otros.9,11–13 Diversos trabajos llaman la atención sobre el papel de algunos teratógenos en la presentación de la secuencia de Moebius, sobre todo de la ingesta de misoprostol en el primer mes dela gestación.9,14–16 Mckusick17 hace la observación de que en 1957, van der Wiel reportó 46 afectados en seis generaciones, y Fortanier 15 casos en tres generaciones; observándose, en ambos estudios, un patrón de distribución vertical, concordante con la herencia autosómica dominante. Becker y Lund,18 documentaron 15 casos en una familia, en la cual encontraron consanguinidad entre los progenitores, y en donde la enfermedad respetó a los padres pero afectó a hijos de ambos sexos, sugiriendo un patrón de herencia autosómica recesiva.
Diversos reportes señalan el involucro de aberraciones cromosómicas de tipo estructural: Ziter y col.,19 en 1977, observaron en el cariotipo de siete afectados de tres generaciones de una misma familia, la presencia en cada uno de ellos de una translocación recíproca entre los cromosoma 1 y 13, teniendo como puntos de rompimiento la región 3–4 del brazo corto del cromosoma 1, y la región 1–3 del brazo largo del cromosoma 13, sugiriendo que el locus para el gene de la secuencia de Moebius pudiera encontrarse en el brazo corto del cromosoma 1, o en el brazo largo del cromosoma 13.20 Recientemente, Hedges y col.21 corroboraron estos hallazgos.
En el presente estudio, 100% de los casos presentaron inexpresividad facial, existiendo en todos los pacientes disfunción del habla. Con relación a lo considerado por Cronemberger y col.,1 en el presente estudio, 23% manifestaron alteración de las extremidades, consistente en pie equino varo. Los padecimientos asociados al síndrome de Moebius que se encontraron fueron: secuencia de Poland, secuencia de Pierre Robin, malformación anorrectal y labio hendido. Los rayos "X" (radiografía simple de abdomen en el segundo trimestre del embarazo) y la ingesta de misoprostol en el segundo mes de embarazo, fueron los teratógenos que se logró documentar en dos pacientes. Todos los casos en el presente estudio fueron esporádicos y en 11 se realizaron los análisis cromosómicos con resultados de normalidad de los mismos.
El límite de edad de los pacientes que asisten al Hospital del Niño "Dr. Rodolfo Nieto Padrón" en Tabasco, México, es de 0 a 14 años y el seguimiento en el Servicio de Genética se da durante los primeros cinco años de la vida, edad en que pueden aparecer complicaciones asociadas.
Los autores del presente trabajo consideran que el diagnóstico temprano de la secuencia de Moebius favorece la planeación en el manejo del enfermo, y al mismo tiempo ofrece a los padres de los afectados un asesoramiento genético oportuno.
Referencias
1. Cronemberger MF, de Castro–Moreira JB, Brunoni D. Ocular and clinical manifestations of Moebius syndrome. J Pediatr Ophthalmol Strabismus. 2001; 38: 156–62. [ Links ]
2. Villafranca AJ, Castillo DP, Garcés SM, Villalón FE, Grez LE, Díaz GA. Síndrome de Moebius. Rev Chilena Cir. 2003; 55: 75–80. [ Links ]
3. Terzis JK, Noah EM. Dynamic restoration in Moebius and Moebius–like patients. Plast Reconstr Surg. 2003; 111: 40–55. [ Links ]
4. Barba PSA, Camacho PMA, Ramírez AC, Cano JGZ, Ávila FF. Parálisis facial congénita. Reportedeun caso. An Radiol Mex. 2006; 4: 337–43. [ Links ]
5. Stromland K, Sjogreen L, Millar M. Moebius sequence: a Swedish multidiscipline study. Eur J Paediatr Neurol. 2002; 6: 35–45. [ Links ]
6. Momtchilova M, Pelosse B, Rocher F, Renault F, Laroche L. Moebius syndrome: ocular and clinical manifestations. J Fr Ophtalmol. 2007; 30: 177–82. [ Links ]
7. Verzil HT, van der Zwaag B, Cruysberg JR, Padberg GW. Moebius syndrome redefined: a syndrome of rhombencephalic mal development. Neurology. 2003; 61: 327–33. [ Links ]
8. Dooley JM, Stewart WA, Hayden JD, Therrien A. Brainstem calcification in Moebius syndrome. Pediatr Neurol. 2004; 30: 39–41. [ Links ]
9. Miller MT, Strömland K, Ventura L, Johansson M, Bandim JM, Gillberg C. Autism associated with conditions characterized by developmental errors in early embryogenesis: a mini review. Int J Dev Neurosci. 2005; 23: 201–19. [ Links ]
10. Issaivanan M, Virdi VS, Parmar VR. Subclavian artery supply disruption sequence Klippel–Feil and Moebius anomalies. Indian J Pediatr. 2002; 69: 441–2. [ Links ]
11. Yeh PC, Kipp MA. A case of Moebius syndrome in association with klinefelter syndrome. Ophthalmic Genet. 2002; 23: 185–9. [ Links ]
12. Verloes A, Bitoun P, Heuskin A. Moebius sequence, Robin complex, and hypotonia: severe expression of brainstem disruption spectrum versus Carey–Fineman–Ziter syndrome. Am J Med Genet. 2004; 127: 294–7. [ Links ]
13. Felice KJ, Jones JM, Conway SR. Facioscapulaohumeral dystrophy presenting as infantile facial diplegia and late–onset limb–girdle myopathy in members of the same family. Muscle Nerve. 2005; 32: 368–72. [ Links ]
14. Da Silva dal Pizzol T, Knop FP. Prenatal exposure to misoprostol and congenital anomalies: systematic review and meta–analysis. Reprod Toxicol. 2006; 22: 666–71. [ Links ]
15. Marques–Díaz MJ, González CH, Rosemberg S. Moebius sequence in children exposed in utero to misoprostol: neuropathological study of three cases. Birth Defects Res A Clin Mol Teratol. 2003; 67: 1002–7. [ Links ]
16. Sánchez O, Guerra D. Moebius syndrome due to the use of misoprostol. Case report. Invest Clin. 2003; 44: 147–53. [ Links ]
17. McKusick VA. Moebius syndrome. Online Mendelian inheritance in man. 2003. Disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=157900. [ Links ]
18. Becker CF, Lund HT. A family with Moebius syndrome. J Pediatr. 1974; 84: 115–7. [ Links ]
19. Ziter FA, Wiser WC, Robinson A. Three generation pedigree of a Moebius syndrome variant with chromosome translocation. Arch Neurol. 1977; 34: 437–42. [ Links ]
20. Johansson M, Wentz E, Fermell E. Austistic spectrum disorders in Moebius sequence: a comprehensive study of 25 individuals. Dev Med Child Neurol. 2001; 43: 338–45. [ Links ]
21. Hedges DW, Jeppson KG, Burns C. Twenty–year behavioral follow–up of a 1;13 chromosomal translocation and Moebius syndrome presenting with poor impulse control, exhibitionism, and aggression. Compr Psychiatry. 2003; 44: 462–5. [ Links ]

