










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.65 no.4 México jul./ago. 2008
Caso clínico patológico
Lactante con hepatoesplenomegalia masiva
Infant with great hepatosplenomegaly
Guillermo Ramón–García1, Bertha Soria–Garibay2
Departamento de Patología
Hospitalización, Hospital Infantil de México Federico Gómez, México, D.F., México
Solicitud de sobretiros:
Dr. Guillermo Ramón García
Depto. de Patología,
Hospital Infantil de México Federico Gómez,
Calle Dr. Márquez Núm. 162, Col. Doctores,
Deleg. Cuauhtémoc, C.P. 06720, México, D.F., México.
Fecha de recepción: 15–05–2008.
Fecha de aprobación: 04–06–2008.
Resumen de la historia clínica
(A 0628)
Paciente femenina de 14 meses de edad. Motivo de consulta: palidez, hiporexia y fiebre. Antecedentes heredofamiliares: madre de 20 años sana, padre de 20 años ayudante de albañil con alcoholismo positivo, sano. Una hermana de dos años cinco meses sana. Resto sin importancia. Antecedentes no patológicos: originarios y residentes del Estado de México, nivel socioeconómico bajo. Alimentado al seno materno hasta su ingreso, ablactado a los tres meses de edad con frutas y verduras. Inmunizaciones completas. Antecedentes perinatales: producto de la gesta 2, embarazo, parto y período perinatal sin complicaciones. Peso al nacimiento de 3 300 g, valoración de Apgar se desconoce.
Padecimiento actual: inició 20 días previos a su ingreso (2 de mayo de 2006) con hiporexia y palidez. A los cinco días se le agregó fiebre no cuantificada, de predominio nocturno, sin escalofríos o diaforesis y que cedía con medios físicos y antipiréticos.
Exploración física: peso 8 kg, talla 74 cm, frecuencia cardiaca (FC) 140/min, frecuencia respiratoria (FR) 44/min, tensión arterial (TA) 90/60 mm Hg, temperatura 37.2° C, perímetro abdominal 47 cm, perímetro cefálico 44 cm; activa, sin facies característica, cabeza y cuello sin alteraciones, no se apreciaron adenomegalias. Tórax sin anormalidades; abdomen globoso a expensas de hígado a 4–5–3 cm debajo del borde costal derecho, bazo a 5 cm; adenomegalia de 1 cm de diámetro, móvil, en región inguinal.
Se le realizó un aspirado de médula ósea que mostró datos de proceso reactivo sin evidencia de infiltración neoplásica. Hemocultivo positivo para bacterias grampositivas, Comamonas acidovorans. Se le dio manejo con cefotaxima a 150 mg/kg/ día. Tomografía axial computada (TAC) de tórax y abdomen con adenomegalias hiliares, hepáticas y esplénicas. Un nuevo aspirado–biopsia de médula ósea mostró hiperplasia de la serie roja; megacariocitos aumentados, numerosos monocitos e histiocitos activados, y solo uno con hemofagocitosis. Mielocultivo negativo. Los resultados de los exámenes de laboratorio se muestran en el cuadro 1.
A los 15 días de su ingreso (6 de junio de 2006) se le realizó, mediante laparoscopia, biopsia de hígado y bazo que mostraron infiltración difusa por histiocitos; sin embargo, no fue posible llegar a un diagnóstico por microscopia de luz. A los 26 días de estancia hospitalaria un hemocultivo central fue positivo a bacilos gramnegativos, con aislamiento de Salmonella grupo D; el cultivo de punta de catéter mostró más de 100 000 UFC de Staphylococcus epidermidis. Se inició manejo con vancomicina a 40 mg/kg/día por 14 días.
El 4 de julio de 2006 se integró diagnóstico de síndrome hemofagocítico asociado a infección. Se descartaron alteraciones lisosomales (beta glucosidasa, quitiotriosidada y esfingomielinasa normales). Se indicó un ciclo de gammaglobulina intravenosa a 400 mg/kg/día por cinco días. El 5 de julio el resultado de las reacciones de inmunohis–toquímica, realizadas en el tejido hepático y esplénico, mostró infiltración histiocítica con positividad para S100 y Cd 1a. El 13 de julio, con base a los hallazgos previos, se consideró el diagnóstico de histiocitosis de células de Langerhans de alto riesgo por la disfunción hepática, hematopoyética y esplénica. Se inició tratamiento a base de etopósido, vinblastina y dexametasona, con los resultados de laboratorio que se muestran en el cuadro 2.
Se le transfundió concentrado plaquetario, observándose mejoría, por lo que se egresó el 18 de julio de 2006. Cuatro días después recibió quimioterapia ambulatoria con vincristina, acudiendo a Urgencias el día 25 de julio con datos de desequilibrio hidroelectrolítico por cinco evacuaciones disminuidas de consistencia, sin moco ni sangre, con llanto sin lágrimas, mucosa oral seca, faringe hiperémica. Se dio tratamiento con plan A de hidratación oral y posteriormente fue egresada a las 13 horas. Acudió finalmente a las 23 horas con ausencia del esfuerzo respiratorio por 30 min, pálida, sin pulsos, hipotérmica, con rigidez muscular generalizada.
Discusión del caso clínico
Dra. Bertha Soria Garibay (Médico Adscrito a Hospitalización). El caso que nos ocupa hoy es el de una lactante de 12 meses de edad con hepatoesplenomegalia, uno de los mayores retos en pediatría. Como en cualquier paciente, los antecedentes son de mucha importancia para encontrar pistas que nos ayuden a solucionar los problemas que se nos presentan en la evaluación de nuestros pacientes. Ante un niño con hepatoesplenomegalia, es importante buscar antecedentes dedaño hepático previo como ictericia, prurito, coluria, acolia, aparición de equimosis con facilidad, eventos de hipoglucemia quizá, si queremos descartar hepatopatía crónica con hipertensión porta, hematoquezia, hematemesis o melena. También son importantes síntomas como falla en el medro, vómito o diarrea crónica, así como pérdida de habilidades adquiridas. Es importante conocer antecedentes de pérdidas fetales y neonatales inexplicables, antecedentes de enfermedades degenerativas o neurológicas progresivas que nos ayuden a descartar enfermedades metabólicas. Si queremos descartar procesos infecciosos, es importante descartar antecedentes epidemiológicos de contactos con animales de granja, domésticos con personas enfermas de hepatitis, tuberculosis, viajes recientes o residencia, ingesta de medicamentos o tóxicos. En este caso, se trataba de una lactante originaria y residente de Chalco, Estado de México, producto de la gesta 2, de padres jóvenes, con una hermana en edad preescolar, con antecedentes de infecciones de vías aéreas superiores, que a esta edad son frecuentes, sin nada más relevante. El embarazo no fue con control prenatal regular; sin embargo, la paciente nació en medio hospitalario con buen peso; no contamos con la valoración de Apgar, aunque no hay datos en la historia que sugieran que hubo asfixia, además de que refieren que hubo llanto al nacer y fue egresada con la madre sin complicaciones. La alimentación y el desarrollo psicomotor, a pesar de que la información con que contamos es precaria, sugiere que fue normal. El esquema de vacunación estaba completo. Su padecimiento actual fue de 20 días previos a su ingreso, con un cuadro que se caracterizó por palidez progresiva, hiporexia y fiebre, esta última de predominio nocturno y que se controlaba con antipiréticos y con medicamentos no especificados en la historia clínica. A la exploración física a su ingreso, a la simple inspección no se refería ictericia, estigmas de hepatopatía crónica, o ascitis, sólo llamaba la atención la presencia de abdomen prominente. La somatometría reflejó un déficit de 15% para el peso, sin déficit de talla para la edad, lo que apoyaba una desnutrición de primer grado secundaria a una afección aguda. Se registró también un síndrome anémico, sustentado por la palidez, la taquicardia y el soplo cardiaco, dato que corroboramos después con una cifra de hemoglobina (Hb) de 5.1 mg/dL. Se integró también al expediente un síndrome infiltrativo por la presencia de hepatomegalia y esplenomegalia masiva (se entiende por esplenomegalia masiva cuando el bazo llega a fosa iliaca o sobrepasa la línea media), además de adenomegalia inguinal izquierda. Los exámenes de laboratorio, además de la anemia, muestran trombocitopenia e hipoalbuminemia de 2.5 mg/dL. El abordaje de un paciente con hepatoesplenomegalia es muy extenso; sin embargo, ante un paciente con estas características, y con fiebre, estamos obligados a considerar dos categorías, la infecciosa y la infiltrativa. Aunque existen distintas entidades en las que estas manifestaciones pueden combinarse, sólo en algunas se presentan simultáneamente. Además, el hecho de diferenciar un proceso infeccioso de un infiltrativo, en un lactante sintomático, constituye una urgencia. Debido a que tanto el hígado como el bazo constituyen parte del sistema retículo–endotelial, la causa más frecuente de crecimiento de los mismos es la respuesta inflamatoria ante un proceso infeccioso. Por estadística, los procesos infecciosos más frecuentes son los virales. Por eso era importante descartar una infección viral sistémica por virus de Epstein–Barr (VEB), citomegalovirus (CMV), virus de inmunodeficiencia humana (VIH), y también por virus hepatotrópicos. Se ha descrito hepatomegalia en algunas infecciones bacterianas, entre las que destacan salmonelosis y brucelosis; había que descartar tuberculosis y algunas otras entidades como: paludismo, leishmaniasis, toxoplasmosis, histoplasmosis, toxocariasis, esquistosomiasis y leptospirosis, entre otras. De todas estas entidades, las que se presentan con esplenomegalia masiva son: tuberculosis, leishmaniasis y paludismo. No se cuenta con antecedente epidemiológico para ninguna de ellas. En este paciente se descartó Salmonella, Brucela, infección por CMV, paludismo, infección por VEB, virus de la hepatitis B y C, toxocariasis, y VIH. Me llamó la atención la búsqueda intencionada para Toxocara, ya que es una entidad muy rara y no existe antecedente para la misma. Y que no se haya hecho abordaje para tuberculosis, la cual es más frecuente. En términos generales, podemos decir que con todos los exámenes efectuados no se había podido documentar enfermedad infecciosa. Por otra parte, siempre que nos enfrentamos con un lactante con fiebre, hepatoespleno–megalia y bicitopenia, aún sin contar con el hallazgo de blastos en sangre periférica, nos preocupa que se trate de una malignidad. En este grupo de edad, las neoplasias más frecuentes son: leucemia, linfoma, histiocitosis, neuroblastoma y tumor de Wilms. Los tumores hepáticos, aunque raros, son causa frecuente de hepatomegalia y, dependiendo del estadio clínico al momento del diagnóstico, pueden o no acompañarse de esplenomegalia. Sin embargo, la esplenomegalia masiva sólo se encuentra descrita en la histiocitosis y en la leucemia mieloide crónica, la cual es una entidad rara a esta edad. La presencia de cualquier citopenia puede explicarse por disfunción medular por cualquier infiltración; la fiebre en estos casos forma parte del cuadro clínico inicial. En la historia clínica no se describe una radiografía de tórax, estudio muy importante para descartar marca mediastinal, la cual no e staba presente. Dentro del abordaje de fiebre en estudio y bicitopenia, es importante la realización de aspirado de médula ósea para, además de descartar un proceso neoplásico, identificar procesos infecciosos y enfermedades metabólicas mediante tinciones y cultivos. A esta paciente se le realizaron dos, uno a su ingreso y otros 16 días después, además de una biopsia de hueso, en la que no se observaron datos de infiltración maligna, las tres líneas celulares estaban conservadas, aclarando que no todas las enfermedades infiltrativas pueden demostrarse en médula ósea, como en la histiocitosis y linfoma. Los resultados de los exámenes a su ingreso no fueron concluyentes, ni para causas infecciosas ni para infiltrativas. No se observaron blastos en el frotis, no había lisis tumoral, la fosfatasa alcalina y la DHL se encontraban dentro de límites normales, los aspirados y biopsias de médula ósea fueron negativos. Las pruebas serológicas, cultivos y pruebas rápidas para microorganismos también negativos. Contamos con estudios de gabinete realizados en los primeros 15 días, entre ellos la TAC toracoab–dominal, que no mostraron masas mediastinales, en pulmones ni en tórax, ni conglomerados ganglionares. Tampoco se evidenciaron tumores renales ni hepáticos. El gammagrama hepatoesplénico descartó que las bicitopenias hubieran sido por hiperesplenismo. La descripción de la captación del fármaco–trazador en el hígado hubiera sido de utilidad para descartar procesos infiltrativos vs neoplásicos. En la ultrasonografía llama la atención las diferencias de econocidad en el bazo y el hígado, y con el Doppler se descartó hipertensión portal. Las radiografías de cráneo y huesos estaban dentro de lo normal. Se le realizó una biopsia de piel, que si bien hasta un tercio de los pacientes con histiocitosis tiene afección cutánea, a la exploración física la piel de esta paciente era sana, por lo que la biopsia no estaba indicada, y el resultado, como era de esperarse, fue normal. Hasta este momento no teníamos un diagnóstico claro con esta paciente, por lo que debíamos ser más invasivos, realizándose exploración con toma de biopsia hepática y esplénica. A mes y medio de su ingreso, la evolución desde el punto de vista hematológico era desfavorable, siempre con tendencia a la anemia sin demostrar hemólisis, sangrados macro o microscópicos; se le realizó una panendoscopia, a pesar de que no tenía evidencia de hipertensión portal, la posibilidad de hiperesplenismo también fue descartada como ya se había comentado. Era evidente la afección hepática con hipoalbuminemia, se agregó colestasis y tiempo de protrombina alargado. Durante su estancia hospitalaria cursó con algunos procesos infecciosos inherentes tanto a la hospitalización prolongada como a la invasividad a que fue sometida, como catéteres, toma de productos, etc. Con aislamiento de varios gérmenes, entre ellos Pseudomonas aeruginosa, Salmonella del grupo D, y S. epidermidis los cuales fueron tratados en su oportunidad sin mayor trascendencia. La manera en que la paciente seguía evolucionando, sugirió cada vez más histiocitosis. Sin embargo, el reporte fue el de una enfermedad por atesoramiento del tipo Nieman–Pick vs enfermedad de Gaucher. Se descartó que hubiera hemofagocitosis en esas biopsias. Las formas más frecuentes de enfermedades por atesoramiento a esta edad son Nieman–Pick, Gaucher, glucogenosis y mucopolisacaridosis. En todas ellas hay hepatomegalia, pero esplenomegalia masiva solo se describe en Nieman–Pick y Gaucher, siendo más constante en la enfermedad de Gaucher. Existen cinco variedades clínicas de Nieman–Pick, la que comparte más datos con nuestro paciente sería la de tipo B, que se caracteriza por hepatoes–plenomegalia, retardo en el crecimiento, citope–nias e involucro pulmonar, y hasta en 50% la presencia de la mancha rojo cereza en la retina. El crecimiento visceral suele ser lento y progresivo, además de que la edad de presentación es al final de la infancia y al entrar la adolescencia, datos que no concuerdan con nuestra paciente, ni el retardo en el crecimiento, que no se observó en este caso. En la enfermedad de Gaucher se han descrito tres fenotipos; en este caso seria tipo 1 ó no neuropática, que cursa con hepatoesplenomegalia, citopenias, y puede haber involucro músculo–esquelético, caracterizada por lesiones que en imágenes se denomina en matraz de Ehler–Mayer en huesos largos, dato que no tenía la paciente; se refieren también crisis óseas con dolor óseo muy importante, secundario a infiltración a la médula ósea por células de Gaucher. La enfermedad de Gaucher es una posibilidad a considerar, siempre que el paciente tenga esplenomegalia masiva; sin embargo, raramente se acompaña de fiebre y, como en la enfermedad de Nieman–Pick, el curso tan agudo no la apoya. De cualquier manera, ante el reporte histopatológico teníamos que descartarlas. Las determinaciones de enzimas lisosomales beta galactosidasa para Gaucher y esfingomielinasa para Nieman–Pick fueron normales. En una valoración del servicio de Hematología se sugirió un proceso de hemofagocitosis. A la paciente se le dio el beneficio de la duda y se le administró gammaglobulina. No tenemos determinaciones de ferritina, fibrinógeno, y los triglicéridos eran normales. Finalmente, se recibió el reporte complementario de la biopsia hepática y esplénica con diagnóstico compatible con histiocitosis de células de Langerhans con positividad franca para proteína S 100 y Cd1a. Se catalogó como una histiocitosis de alto riesgo, según los criterios de Lahey, por ser menor de 10 años, afección hematológica con Hb menor a 10 g/dL, plaquetas menores a 100 000/mm3, y disfunción hepática apoyada con albúmina menor a 2.5, y bilirrubinas totales arriba de 2.5 mg/dL. La afección esplénica no es un criterio para catalogar como de alto riesgo a la histiocitosis. No se describe la presencia de tos o taquipnea, que sugieran disfunción pulmonar. De haberlos presentado era preciso haber realizado una TAC para descartar involucro pulmonar. No se cuenta con determinaciones de electrolitos séricos, ni balance hídrico, EE urinario o densidad urinaria, fuera de las de su ingreso, para descartar diabetes insípida, que se presenta en 8% de los casos. Y aunque los niños con diabetes insípida tengan lesiones demostradas radiológicamente en estudios de cráneo, en algunos casos pueden estar no presentes. El pronóstico en pacientes con histiocitosis diseminada, o sea aquella que involucra a dos o más órganos, es de 50% a los cinco años. Se inició esquema con quimioterapia con el protocolo dos de la sociedad del Histiocito con prednisona, vinblastina y etopósido, administrándole a esta paciente dexametasona, con evolución favorable en relación a la fiebre, pero no así desde el punto de vista hematológico, ya que persistía con anemia, trombocitopenia y leucopenia grave. Los leucocitos, que se habían mantenido, descendieron hasta 1 100/mm3, llegando a tener cuentas de neutrófilos totales debajo de 1 000/mm3. El descenso de las tres series se considera como actividad tumoral. Tres días después de la última sesión de quimioterapia, acudió al servicio de urgencias con un cuadro de cinco días de evolución, caracterizado por cinco evacuaciones disminuidas de consistencia, sin moco ni sangre, fiebre ni dolor abdominal, con náuseas sin llegar al vómito. A su ingreso se encontraba normotensa, taquicárdica, polipneica, sin fiebre; neurológicamente estaba íntegra, con llenado capilar de 2 seg, mucosa oral seca y llanto con lágrimas. Para mí, existe una discordancia entre la valoración del estado de hidratación y los signos vitales, ya que a pesar de estar afebril, llegó sumamente taquicárdica y taquipneica, lo que apoyaría más para deshidratación grave. Esto se apoya con otros datos como la hemoconcentración, la Hb de ese día era de 15.2 g/dL, tres días antes era de 10.8 g/dL. A pesar de, según la historia, no estar deshidratada, se le dieron dos planes de hidratación con vida suero oral, uno en urgencias y otro en quimioterapia. Esta paciente no debió pasar a la sala de quimioterapia a continuar con su hidratación, sino que debió continuar en la sala de urgencias con monitoreo estrecho. El único estudio solicitado fue una biometría hemática, pero no se encontró neutropenia, sino leucocitosis y bandemia. Hubiera sido de mucha utilidad conocer determinaciones de electrolitos séricos, bicarbonato y densidad urinaria.
Se debieron haber hecho cuatro cosas básicamente: 1. Descartar alteraciones electrolíticas y del equilibrio ácido base, para clasificarla como deshidratación grave y normar la conducta, valorando si ameritaba hidratación endovenosa, incluso descartar diabetes insípida, ya que el cuadro enteral no era de tal magnitud; 2. Descartar íleo por vincristina, exacerbado por alteraciones electrolíticas; 3. Descartar proceso infeccioso por alteraciones encontradas en la biometría hemática y por ser una paciente de riesgo; y 4. Debió haber permanecido por lo menos 24 horas en observación, hasta asegurar un adecuado estado de hidratación, con adiestramiento a la madre. No obstante, se egresó a la paciente a su domicilio con tratamiento de trimetoprim–sulfametoxazol a dosis subterapéuticas, sin quedar clarolaindicación del medicamento, ni el por qué de esta dosis; 10 horas después regresan los padres al hospital con la niña muerta. Es importante puntualizar que nuestra responsabilidad debe ir más allá del diagnóstico y tratamiento. Debe extenderse a los efectos de la terapéutica que empleamos a corto, mediano o largo plazo; considerarán que la enfermedad y las características de nuestras pacientes les agrega una comorbilidad muy importante, por eso siempre tenemos que manejarlas como pacientes de alto riesgo ante cualquier proceso agregado. La neutropenia no es la única indicación de hospitalización en un paciente oncológico.
Mis diagnósticos finales son: histiocitosis de células de Langerhans, diseminada de alto riesgo.
Gastroenteritis probablemente infecciosa con deshidratación grave.
Como causa probable de la muerte: choque hipovolémico y desequilibrio hidroelectrolítico.
Hallazgos anatomopatológicos
Dr. Guillermo Ramón García (Departamento de Patología). En las biopsias de hígado se observó denso infiltrado linfohistiocitario en espacios porta, algunos histiocitos mostraban abundante citoplasma de aspecto espumoso, por eso fue que inicialmente se considerara como enfermedad por almacenamiento, pero en los estudios de inmuno–histoquímica se observó que eran positivos para proteína S–100 y CD1a, con lo que se realizó el diagnóstico de histiocitosis de células de Langer–hans (Figs. 1 y 2). En la biopsia de bazo también se encontraron grupos de histiocitos con las mismas características.
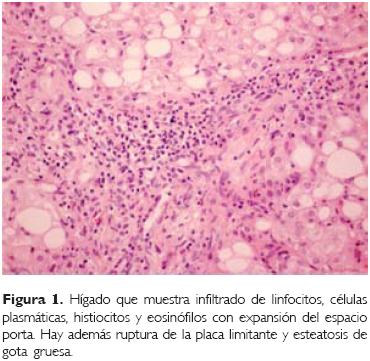

En la autopsia, se observaron datos de desnutrición con peso de 7 800 g vs 9 320 g, pero la talla estaba conservada (71 cm). El hígado aún se encontraba aumentado de tamaño (500 g vs 304 g) (Fig. 3), así como el bazo (108 g vs 26 g), y se encontraron adenomegalias múltiples en mesenterio, retroperitoneales, mediastino y cuello. En todos elloshabía grupos de histiocitos de Langer–hans (Fig. 4). El hígado, además, mostraba esteatosis difusa de gota gruesa. La médula ósea era hipocelular, por efecto de la quimioterapia, pero no había células neoplásicas. Había dilatación de las asas del intestino delgado y grueso, la mucosa mostraba pequeñas zonas de necrosis y ulceración, y leve incremento del infiltrado inflamatorio linfocitario acompañado de edema y congestión. En la lámina propia había, además, bandas de contracción (Figs. 5 y 6). Estos datossonde enterocolitis aguda, probablemente infecciosa, más que de enterocolitis neutropénica. Secundario a esta enterocolitis se encontraron datos de deshidratación, manifestados por dilatación y retención de secreciones en los conductos de las glándulas mucosas del aparato respiratorio, glándulas salivales y páncreas (Fig.7). Tambiénse observaron datos de choque, manifestado en pulmones como daño alveolar difuso con edema alveolar con membra–nashialinas,congestión capilar y hemorragia focal (Fig. 8), y en riñones necrosis tubular aguda. En el sistema nervioso central había datos de atrofia leve, el encéfalo pesó 750 g vs 1 000 g. Microscópicamente se encontró datos de encefalopatía hipóxica aguda, pero lo más llamativo fue una degeneración espongiforme de la sustancia blanca periacueductal, probablemente secundaria a la quimioterapia (Fig. 9).







Los diagnósticos anatomopatológicos finales son:
Enfermedad principal: histiocitosisdecélulas de Langerhans, tratada con quimioterapia (vincristina) con infiltración a hígado, bazo y ganglios linfáticos.
Alteraciones concomitantes: hepatomegalia (500 g vs 304 g).
Esteatosis difusa de gota gruesa.
Esplenomegalia (108 g vs 26 g).
Enterocolitis aguda probablemente infecciosa.
Datos anatómicos de deshidratación en glándulas mucosas del aparato respiratorio y digestivo.
Datos anatómicos de choque manifestado por daño alveolar difuso y miopatía visceral.
Desnutrición.
Atrofia cerebral (750 g vs 1 000 g).
Degeneración espongiforme de sustancia blanca periacueductal probablemente secundaria a quimioterapia.
Causa de la muerte: choque probablemente hipovolémico por deshidratación.
Comentarios
En esta paciente el diagnóstico histopatológico de histiocitosis no se realizó inicialmente, debido a que la infiltración al hígado y bazo como manifestación inicial no es la habitual en este padecimiento; se refiere un caso semejante a obstrucción ya que existe infiltración a los conductos biliares.1 La evolución a la cirrosis puede ocurrir como una complicación debido a la obstrucción, por lo que se ha propuesto al trasplante hepático como tratamiento cuando se presenta la disfunción hepática.2 En el hígado, además del daño a los conductos, se puede también observar infiltración portal y al lobulillo como finalmente se demostró en la autopsia. En la biopsia inicial no estuvieron presentes los cúmulos de células de Langerhans en el lobulillo y sólo habían escasas células grandes en espacios porta, las cuales fueron confundidas como histiocitos con material acumulado pero la inmunohistoquímica finalmente fue clave para el diagnóstico, al demostrarse la positividad para los anticuerpos Cd1a y proteína S–100, los cuales son marcadores de la histiocitosis de células de Langerhans.3,4
Referencias
1. Dehner LP. Morphologic findings in the histiocytic syndromes. Semin Oncol. 1991; 16: 8–17. [ Links ]
2. Favara BE, Jaffe R. Pathology of Langerhans histiocytosis. Hematol Oncol Clin North Am. 1987; 1: 75–9. [ Links ]
3. Leblanc A, Hadchowelt M, Lehan P. Obstructive jaundice in children with histiocytosis X. Gastroenterology. 1981; 80: 134–9. [ Links ]
4. Concepcion W, Esquivel CO, Terry A. Liver transplantation in Langerhans histiocytosis (histiocytosis X). Semin Oncol. 1991; 18: 24–8. [ Links ]

