










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.64 no.2 México mar./abr. 2007
Artículo original
Enfermedad de moyamoya
Moyamoya disease
Marco Urrutia–Ruiz, Eduardo Barragán–Pérez, Juan Hernández–Aguilar, Saúl Garza–Morales, Enoe Cruz–Martínez, Feliciano Santana–García, Edil Escobar–Mendoza
Departamento de Neurología, Hospital Infantil de México Federico Gómez, México, D.F., México.
Solicitud de sobretiros:
Marco Urrutia Ruiz, Dpto. de Neurología
Hospital Infantil de México Federico Gómez
Dr. Márquez # 162, Col. Doctores, Deleg. Cuauhtemoc
C P. 06720, México, D. F., México.
Fecha de recepción: 22–03–2006.
Fecha de aprobación: 05–06–2007.
Resumen
Introducción. Objetivo: describir el comportamiento de la enfermedad de moyamoya en niños atendidos en el Hospital Infantil de México Federico Gómez del año 1995 a 2005.
Material y métodos. Estudio retrospectivo y descriptivo. Se revisaron 7 expedientes clínicos de niños con este diagnóstico, los cuales reunieron los criterios establecidos. Se elaboró una hoja de captura, con datos demográficos y personales, manifestaciones clínicas, estudios diagnósticos, tratamiento y evolución. Se analizaron los casos mediante estadística descriptiva.
Resultados. La edad promedio fue de 6.5 años; predominó en mujeres (4). La manifestación clínica de inicio en todos los pacientes fue hemiparesia desproporcionada aguda o subaguda. Se encontró comorbilidad con: síndrome de Down (4), microcefalia (3), y cardiopatías congénitas (2). El diagnóstico se confirmó con angiografía por sustracción digital (5) y angiorresonancia (2). Se practicó arteriodurosinangiosis en 2 casos y angiosinostosis temporal en 1, con resolución del déficit motor sin deterioro posterior. Los restantes 4 pacientes sólo recibieron tratamiento sintomático, con persistencia de la afección motora y presencia de epilepsia.
Conclusión. El diagnóstico de la enfermedad de moyamoya se realiza con angiografía o angiorresonancia magnética. El tratamiento quirúrgico es de elección.
Palabras clave. Enfermedad de moyamoya; manifestaciones clínicas; diagnóstico; tratamiento.
Abstract
Introduction. Objective: Describe the evolution of moyamoya disease in children attended in the Hospital Infantil de Mexico from 1995 to 2005.
Material and methods. Retrospective and descriptive study. We retrospectively reviewed the medical records of 10 children with this diagnosis; 7 with the established criteria; 3 were excluded for not counting with angiographic conventional and angioresonance for their diagnosis. Demographic and personal information, clinical manifestations, diagnostic studies, treatment and evolution were recorded and the cases analyzed using descriptive statistics.
Results. The average age was 6.5 years and 6/7 was females. The clinical manifestation in all patients was acute or subacute hemiparesis. Associated pathologies included: Down's syndrome (4), microcephaly (3), and congenital heart disease (2). The diagnosis was confirmed with angiography (5), magnetic resonance angiography (2). A duro–arterio–synangiosis was practiced to 2 and a temporal angiosinostosis to 1, with resolution of the motor deficit without posterior deterioration. The remaining 4 patients only received symptomatic treatment, with persistence of motor deficit and seizures.
Conclusion. The diagnosis of moyamoya disease continues to be with angiography and angioresonance. When possible, surgical correction is the treatment of choice.
Key words. Moyamoya's disease; clinical manifestations; diagnosis; treatment.
Introducción
La enfermedad de moyamoya fue descrita por primera vez en 1957 por Takeuchi y Shimizu.1 El término moyamoya fue introducido por Suzuki y Takaku2 en 1969 por el aspecto angiográfico de la circulación colateral a nivel cerebral, en humo de cigarrillo o fumarola (en japonés "moyamoya"). Se considera una vasculopatía oclusiva cerebral progresiva, caracterizada por estenosis u oclusión de la porción supraselar de la arteria carótida interna, principalmente de la arteria cerebral media y de la arteria cerebral anterior.3 Esta entidad afecta mayormente a la población de origen asiático. En Japón, la incidencia y prevalencia es de 0.35 y 3.16 por 100 000 habitantes, respectivamente.4,5 En un estudio realizado en diferentes hospitales de Estados Unidos de Norteamérica se reporta una incidencia de 86/100 000 personas y que varía de acuerdo a las diferentes etnias, siendo más común en los de origen asiático.6 Es más frecuente en el sexo femenino. El síndrome de Down suele estar asociado a esta entidad.7 La etiología es desconocida,8 sin embargo, se ha mostrado que tiene una base genética, con alteración de los cromosomas 3p, 6q y 17q, más recientemente se identificaron alteraciones en el 8q23 y 12p12.9
La base anatomopatológica de esta entidad consiste en una proliferación de las células musculares lisas y su migración hacia la íntima por mecanismos desconocidos, lo que origina una estenosis arterial, ya sea de la carótida interna o de sus ramas principales (cerebral anterior o media), condicionando la aparición de una red de vascularización anómala basal a expensas de arterias lenticuloestriadas y tálamo perforantes, que da una imagen similar al humo de cigarrillo2 en el estudio angiográfico. El Ministerio de Salud de Japón estableció los criterios diagnósticos en 1979, que consisten en:10,11
1. Cambios esteno–oclusivos en las porciones terminales de las arterias carótidas internas intracraneales y desarrollo de vasos "moyamoya" en la base del cerebro.
2. Estos cambios pueden ser bilaterales. Cuando la lesión es bilateral el diagnóstico de la enfermedad de moyamoya es definida, de otra forma es probable.
Los criterios de exclusión incluyen: arteriosclerosis, enfermedades autoinmunes, tumores cerebrales, encefalopatía ac tínica, enfermedad de Von Recklinghausen, síndrome de Down, etc. Estos criterios fueron propuestos por el Comité de Investigación de la Enfermedad de Moyamoya del Ministerio de Salud de Japón.
Muchos casos de pacientes de moyamoya comunicados a nivel mundial no se basan en estos criterios. Cuando la angio–arquitectura es similar a la enfermedad de moyamoya se le llama síndrome moyamoya.
Enfermedad unilateral y cuasi–moyamoya
A excepción de la enfermedad definida moyamoya, las condiciones clínicas similares no están bien establecidas:
1. Los resultados angiográficos típicos se observan solamente en un lado, pero el lado opuesto es totalmente normal.
2. Los resultados angiográficos típicos se observan bilateralmente, pero se asocia a enfermedades sistémicas mencionadas anteriormente.
Otra condición es:
1. La lesión es unilateral, se llama enfermedad de moyamoya unilateral.
2. Si se asocia con una enfermedad sistémica se llama cuasi–moyamoya (akin–cuasi–moyamoya).
Entre quienes padecen la enfermedad de moyamoya unilateral algunos llegan a presentarla en forma bilateral y debido a que la etiología de la enfermedad unilateral o cuasi–moyamoya no está bien aclarada, se tratan generalmente de manera similar a la enfermedad de moyamoya definida.
El cuadro clínico suele iniciarse en la primera década de la vida y los principales síntomas son ataques isquémicos transitorios, hemiparesia, convulsiones parciales, cefalea migrañosa, movimientos involuntarios como hemicorea, en ocasiones como manifestación inicial.12–17 El diagnóstico se sospecha por la clínica y se confirma por los hallazgos de la angiografía o angiorresonancia.18,19 El tratamiento es quirúrgico, y la mayoría de los pacientes mejoraron su sintomatología.20–22 Una de las técnicas quirúrgicas que se ha propuesto como primera opción de tratamiento es la arteriodurosinangiosis, descrita inicialmente por Matsushima y col.,23 y por Matsushima e Inaba,24 posteriormente modificada por los mismos autores. Consiste en realizar una unión estrecha entre la arteria temporal superior con la galea circundante; la modificación consistió en apertura amplia de la duramadre en aposición con la arteria temporal superficial y amplio tejido galeal circundante, con fijación de este tejido a la piamadre de la convexidad cerebral, a la cual se le realizan pequeñas ventanas, y se extirpa la piamadre para lograr un contacto más estrecho entre la arteria donante y el parénquima cerebral.
Material y métodos
Se realizó un estudio retrospectivo de los expedientes clínicos de todos los pacientes con diagnóstico de enfermedad de moyamoya atendidos en el Hospital Infantil de México Federico Gómez en el período entre 1995 y 2005. Se encontró un total de 10 casos, de los cuales sólo siete contaban con resultados de estudios de neuroimagen que permitieron corroborar esta entidad.
Resultados
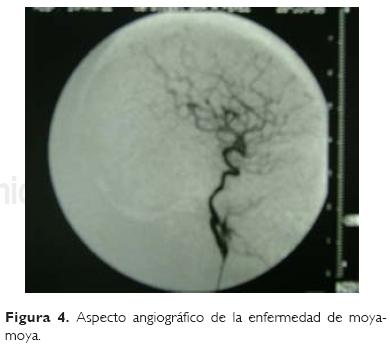
Se revisaron 10 expedientes clínicos de pacientes con diagnóstico de enfermedad de moyamoya en el período 1995–2005, de los cuales sólo siete cumplieron con los criterios clínicos y de neuroimagen, tres se excluyeron por no contar con resultados de angiografía o angiorresonancia que permitieran corroborar el diagnóstico, puesto que es conocido que se trata de una vasculopatía oclusiva o estenótica de las arterias carótida interna, arteria cerebral media o cerebral anterior. El promedio de edad de los pacientes fue de 6.5 años, cinco menores de 10 años y sólo dos mayores de 10 años; el sexo predominante fue el femenino (cuatro). Todos los pacientes debutaron con una hemiparesia desproporcionada aguda o subaguda, siendo más frecuente del lado derecho (cuatro), seguido de convulsiones (cuatro), de los cuales uno presentó crisis tónico–clónico generalizada y el resto crisis parciales hemicorporales (Cuadro 1). La enfermedad más frecuente asociada a esta entidad fue el síndrome de Down (cuatro) y la cardiopatía congénita (dos, uno con comunicación interauricular e interventricular y el otro con estenosis de la pulmonar). El retardo del desarrollo psicomotor se presentó en cinco prematuros, muy probablemente debido al síndrome de Down y a la microcefalia (Fig. 1). El diagnóstico (Fig. 2) se corroboró con angiografía por sustracción digital en cinco pacientes: dos presentaban estenosis de la porción supraclinoidea de la arteria carótida interna (uno unilateral y otro bilateral) y tres estenosis de la arteria cerebral medial (en dos casos el defecto fue unilateral y en uno bilateral). Se practicó angiorresonancia a cuatro, cuyos hallaz.gos fueron: hipoplasia de la carótida interna en uno (bilateral, corroborado con angiografía) y de la cerebral media en tres (dos unilateral y uno bilateral, este último no corroborado por angiografía), por tanto, de los cuatro, en dos el estudio se consideró como diagnóstico definitivo, y en los otros dos se corroboró su diagnóstico con angiografía por sustracción digital (que se incluyen en los cinco de angiografía). A todos se les hizo tomografía axial computada de cráneo, de los cuales seis presentaron infarto isquémico (dos de arteria cerebral anterior y cuatro a nivel del territorio de la cerebral media) y sólo uno hemorrágico (cerebral anterior izquierdo). A cinco se les tomó electroencefalograma de inicio, de los cuales en tres se reportó disfunción cerebral por disminución del voltaje en la zona del infarto, y en los otros dos se reportó actividad epiléptica: uno en regiones centroparietal y temporal derecha, además de disfunción cerebral por menor voltaje en hemisferio derecho y en el otro actividad epiléptica generalizada moderada. En relación al tratamiento quirúrgico, éste se practicó a tres niños, utilizando la técnica quirúrgica de céfalo arterio–durosinangiosis (dos pacientes) con defecto bilateral y la neuroangiosinostosis temporal izquierda en un paciente. No se reportaron complicaciones en ambos procedimientos. Hubo una evolución excelente en un paciente con recuperación total del déficit motor, sin alteración cognitiva, corroborado por test visomotor de Bender y el de motrices progresivas de Raven, además de la valoración clínica. Con relación a la angiografía de control (uno y dos años después) se observó la anastomosis temporal superficial permeable con aumento de la irrigación en la zona afectada. El segundo paciente operado con la técnica arterio–durosinangiosis, presentaba síndrome de Down, y tuvo mejoría de su déficit motor pero manifestó una epilepsia parcial sintomática y fue tratado con fenobarbital a 5 mg/kg/día. En su control angiográfico se encontró un aumento de la irrigación del territorio cerebral afectado, con permeabilidad de la anastomosis realizada e imágenes de infarto antiguo; sin embargo, dos años después del tratamiento quirúrgico desarrolló leucemia linfoblástica aguda y falleció un año más tarde por choque séptico, además de neutropenia y trombocitopenia graves. El tercer paciente con síndrome de Down, operado con la misma técnica bilateral, tuvo mejoría de su déficit motor, pero evolucionó con epilepsia parcial sintomática y trastornos conductuales como hiperactividad, agresividad y dislalia; llevó tratamiento con carbamazepina 15 mg/kg/día y tioridazina por cinco años. En la angiorresonancia de control se reportó infarto antiguo en región fronto–parietal derecha, estenosis de la carótida interna derecha y anastomosis permeable. De los pacientes no tratados quirúrgicamente (cuatro), dos presentaban síndrome de Down, en todos persistió el déficit motor y desarrollaron epilepsia (tres, epilepsia parcial sintomática, y uno con epilepsia generalizada sintomática), corroborados con estudios de electroencefalografía; un paciente presentó además movimientos coreatetósicos. Todos los niños recibieron rehabilitación.



Discusión
Como se sabe, esta entidad suele ser más frecuente en individuos de origen asiático, y menos en los llamados hispanos, como lo muestran los estudios realizados en Japón y Estados Unidos de Norteamérica;4–6 sin embargo, debe sospecharse para poder llegar al diagnóstico, como se refleja en nuestra casuística, donde de un total de 10 pacientes con sospecha de enfermedad de moyamoya, siete cumplían con los criterios clínicos y de imagen. Se pudo corroborar que el grupo de edad predominante fue el de los menores de 10 años, siendo más frecuente en el sexo femenino. Battistella y Carollo,18 en un estudio realizado en Italia con 34 pacientes, encontraron que la mayoría de ellos iniciaron las manifestaciones clínicas antes de los 16 años, en promedio 5.4 años, predominando en las mujeres. Veinte casos tuvieron eventos isquémicos como manifestación inicial. Similar a lo encontrado en nuestra serie (seis).
Con relación a la forma de presentación de esta entidad, en los pacientes de este trabajo la manifestación clínica inicial fue la hemiparesia asociada a infarto isquémico, de la misma manera que lo reportó Kuroda y col.4 en su estudio de 55 casos, de los cuales 32 eran menores de seis años y tenían una incidencia más significativa de infarto isquémico. Otro signo menos frecuentemente observado en nuestros pacientes fue la corea, presente sólo en un caso, donde no constituyó la manifestación inicial. Hong y col.16 sí reportan un caso de hemicorea como manifestación inicial, evidenciando una hipoperfusión cerebral a nivel del territorio de la carótida interna, corroborada por tomografía con emisión de positrones.
Con relación a la enfermedad asociada, está descrito que sufren mayor predisposición a esta entidad aquellos pacientes con síndrome de Down. Andrew y col.,25 en un estudio de 181 pacientes diagnosticados con enfermedad de moyamoya, refieren que 16 (8.8%) tenían síndrome de Down. En nuestra casuística se encontró que cuatro tenían este síndrome.
El diagnóstico de la enfermedad de moyamoya se basa en los parámetros clínicos y radiológicos encontrados (angiografía o angiorresonancia), así lo refieren diferentes autores, como es el caso de Smith y Scott20 y Houkin y col.19 (Figs. 3 y 4). De la misma forma se confirmó en nuestro grupo de pacientes el diagnóstico, a través de las manifes' taciones clínicas y por medio de angiografía por sustracción digital y angiorresonancia. A cinco casos se les hizo el diagnóstico definitivo con angiografía y a dos sólo con resonancia, aunque se sigue considerando la angiografía el estándar de oro para el diagnóstico definitivo, la angiorresonancia ha venido ganando terreno en este sentido, por tratarse de un método no invasivo.19,20 Los hallazgos encontrados fueron en su mayoría unilateral (cinco), siendo sólo en dos pacientes el defecto bilateral, esto difiere de los criterios iniciales propuestos por el Ministerio de Salud de Japón; sin embargo, se ha visto mayor incidencia de casos unilaterales con formación de vasos moyamoya, a los cuales se les ha llamado atípicos, tal como lo reportan otros autores como Calderelli y col.,26 o Quintana,27 donde describen 10 casos de enfermedad de moyamoya en su trabajo de revisión durante 20 años, encontrando que 50% de sus casos fueron atípicos (unilaterales), similar a lo encontrado en nuestra serie (cuatro). A tres de nuestros pacientes se les practicó tratamiento quirúrgico con muy buenos resultados, similar a lo reportado por Han y col.21 en un estudio realizado en Corea, donde los pacientes sometidos a neurocirugía obtuvieron un beneficio de 73% con las técnicas quirúrgicas ya mencionadas.20

Calderelli y col.26 reportaron nueve pacientes tratados quirúrgicamente con la técnica durosinangiosis, seis con defectos bilaterales y dos con defecto unilateral, con muy buenos resultados, refiriendo mejoría de las manifestaciones clínicas e incremento del flujo sanguíneo cerebral en las zonas afectadas.
Como conclusiones se puede mencionar que la enfermedad de moyamoya es una vasculopatía crónica progresiva, que se presenta como eventos vasculares isquémicos y afección motora unilateral en la edad pediátrica, a diferencia del adulto en que se presenta generalmente con evento vascular hemorrágico. El diagnóstico de elección es mediante la angiorresonancia magnética nuclear o angiografía por sustracción digital. El diagnóstico temprano y tratamiento quirúrgico oportuno mejora el pronóstico y evolución de los pacientes.
Referencias
1. Takeuchi K, Simizu K. Hypogenesis of bilateral internal carotid arteries. No To Shinkei. 1957; 9: 37–43. [ Links ]
2. Suzuki J, Takaku A. Cerebrovascular Moya–Moya disease: Disease showing abnormal net like vessel in base of brain. Arch Neurol. 1969; 20: 288–99. [ Links ]
3. Keisure U, Meyer F, Mellinger J. Moya–Moya disease: The disorder and surgical treatment. Mayo Clinic Pro. 1994; 69: 749–57. [ Links ]
4. Kuroda S, Ishikawa T, Houkin K, Nanba R, Hokari M, Iwasaki Y. Incidence and clinical features of disease progression in adult moyamoya disease. Stroke. 2005; 36: 2148–53. [ Links ]
5. Wakai K, Tamakoshi A, Likezaki K, Fukui M, Kawamura I, Aoki E, et al. Epidemiological features of moyamoya disease in Japan: findings from a nationwide survery. Clin Neurol Neurosurg. 1997; 99 (Supl 2): S1–5. [ Links ]
6. Uchino Kl, Johnston SC, Becker KJ, Tirschwell DL. Moyamoya disease in Washington State and California. Neurology. 2005; 65: 956–8. [ Links ]
7. Ergoven M, Deveci M, Turgut T. Moyamoya disease and Down syndrome, Indian J Pediatr. 2005; 72: 697–9. [ Links ]
8. Peña–Tapia PG, Horn P, Schmiedek P. Moyamoya disease. Rev Neurol. 2006; 43: 287–94. [ Links ]
9. Sakurai K, Horiuchi Y, Ikeda H. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J Hum Genet. 2004; 49: 278–81. [ Links ]
10. Ikeda H, Sasaki T, Yoshimoto T, Fujui M, Arinami T. Mapping of a familial moyamoya disease gene to chromosome 3p24–p26. Am J Hum Genet. 1999; 64: 533–7. [ Links ]
11. Fukuyama Y, Imaizumi T, Osawa M. A long–term prognosis of children with TIAtype spontaneous occlusion of the circle of Willis (moyamoya disease). Japan: Annual report 1993, their research committee on spontaneous occlusion of the circle of Willis (Moyamoya disease) of the Ministry of Health and Welfare; 1993. p. 14–7. [ Links ]
12. Galicchio S, Maza E, Jaimovich R, Arroyo H. Enfermedad de Moya–Moya. Arch Arg Pediatr. 1998; 96: 263–7. [ Links ]
13. Kuroda S, Nanba R, Ishikawa T. Clinical manifestation of infantile moyamoya disease. No Shinkei Geka. 2003; 31: 1073–8. [ Links ]
14. Erguven M, Deveci M, Turgut T. Moyamoya disease and Down syndrome. Indian J Pediatr. 2005; 72: 697–9. [ Links ]
15. Im SH, Oh CW, Kwon OK, Cho BK, Chung YS, Han DH. Involuntary movement induced by cerebral ischemia: pathogenesis and surgical outcome. J Neurosurg. 2004; 100: 877–82. [ Links ]
16. Hong YH, Ahn TB, Oh CW, Jeon BS. Hemichorea as an initial manifestation of moyamoya disease: reversible striatal hypoperfusion demonstrated on single photon emission computed tomography. Mov Disord. 2002; 17: 1380–3. [ Links ]
17. de Veber GA. Enfermedad de Moyamoya. Cerebrovascular disease. En: Swaiman, editor. Neurología pediátrica. Principios y prácticas. Vol II. 2006, St. Louis Baltimore, New York: Mosby; 2006. Vol. II. 1772–3. [ Links ]
18. Battistella PA, Carollo C. Clinical and neuroradiological findings of moyamoya disease in Italy. Clin Neurol Neurosurg. 1997; 99 Suppl 2: S54–7. [ Links ]
19. Houkin K, Nakayama N, Kuroda S. Novel magnetic resonance angiography stage grading for Moyamoya disease. Cerebrovasc Dis. 2005; 30: 347–54. [ Links ]
20. Smith ER, Scott RM. Surgical management of moyamoya syndrome. Skull Base. 2005; 15: 15–26. [ Links ]
21. Han DH, Kwon OK, Byun BJ. A cooperative study: Clinical characteristics of 334 Korean patients with moyamoya disease treated at neurosurgical institutes (1976–1994). The Korean Society for Cerebrovascular Disease. Acta Neurochir(Wien). 2000; 142: 1263–74. [ Links ]
22. Fung IW, Thompson D, Ganesan V. Revascularization surgery for paediatric moyamoya: a review of the literature. Child Nerv Syst. 2005; 21: 358–64. [ Links ]
23. Matsushima Y, Fukai M, Tanaka K. A new surgical treatment of moyamoya disease in children: a preliminary report. Surg Neurol. 1981; 15:313–20. [ Links ]
24. Matsushima Y, Inaba Y. Moyamoya disease in children and its surgical treatment. Introduction of a new surgical procedure and its follow–up angiograms. Child Brian. 1984; II: 155–70. [ Links ]
25. Jea A, Smith ER, Robertson R, Scott RM. Moyamoya syndrome associated with Down syndrome outcome after surgical revascularization. Pediatrics. 2005; 116: 694–701. [ Links ]
26. Calderelli M, Di Rocco C, Gaglini P. Surgical treatment of moyamoya disease in pediatric age. J Neurosurg Sci. 2001; 24: 85–91. [ Links ]
27. Quintana M. Experiencia de 20 años en el manejo de la enfermedad de moyamoya. Rev Chilena Neurocir. 2004; 37: 74–82. [ Links ]

