










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.61 no.1 México feb. 2004
Bol Méd Hosp Infant Méx 2004; Vol. 61(1):68-72
CASO CLÍNICO
Síndrome de Pfeiffer tipo 2 con expresividad variable
Dr. Luis Gómez Valencia
Servicio de Genética
Quim. Anastasia Morales Hernández
Quim. Josefina Salomón Cruz
Laboratorio de Citogenética
Dr. Alfonso Jesús Berttolini Díaz
Servicio de Ortopedia y Traumatología
Dr. Ezequiel Toledo Ocampo
Servicio de Medicina Preventiva
Dr. Ramón Miguel Cornelio García
Servicio de Anestesiología, Hospital del Niño Dr. Rodolfo Nieto Padrón, Villahermosa, Tabasco, México.
Solicitud de sobretiros: Dr. Luis Gómez Valencia, Servicio de Genética, Hospital del Niño Dr. Rodolfo Nieto Padrón, Avenida Gregorio Méndez Magaña No. 2832, Col. Tamulté, Villahermosa, Tabasco, México.
Fecha de recepción: 04-04-2003.
Fecha de aprobación: 30-10-2003.
Resumen
Introducción. Objetivo: presentar un caso clínico de síndrome de Pfeiffer, de 5 años de edad, con cráneo en trébol, proptosis ocular severa, y aparentemente sin retardo mental
Caso clínico. Niño de 5 años de edad, producto de segunda gesta, embarazo normoevolutivo de término; padres de 19 años de edad al momento de nacer el propositus, sin antecedentes teratogénicos, ni consanguinidad o de otro padecimiento similar en miembros de la familia. A la exploración física: cráneo en trébol, frente amplia y prominente, proptosis ocular (antecedente de salida del globo ocular derecho en dos ocasiones), aplanamiento medio facial, pabellones auriculares con hélix de configuraciones en cruz horizontal, primer dedo de manos y pies anchos de su falange distal, sindactilia parcial en manos y pies. El cariotipo en linfocitos de sangre periférica mostró un complemento cromosómico normal 46, XY. Radiológicamente se observó cráneo en trébol, con múltiples impresiones digitales.
Conclusión. El caso presentado aquí corresponde clínica y radiológicamente a un síndrome de Pfeiffer tipo 2, sin complicaciones viscerales y con desarrollo neurológico de acuerdo a su edad cronológica.
PALABRAS CLAVE:Síndrome de Pfeiffer; cráneo-estenosis; cráneo en trébol; cráneo-sinostosis; acro-céfalo-sindactilia.
ABSTRACT
Introduction. Since 1964, about 30 cases of Pfeiffer syndrome type 2 have been informed; this variant is characterized by cloverleaf skull, prominent forehead, severe ocular proptosis, severe central nervous system damage, elbow synostosis, and early death.
Case report. A 5 years old male withouth antecedents of consanguinity, teratogenic exposure of his parents (of 19 years of age at the time of the bird of patient), or familial malformation, was admitted. On physical examination a cloverleaf skull, wide forehead, ocular proptosis (in 2 previous occasions the right eye exited), mid facial flattening, horizontal cross configuration of ear helix, widening of the first finger of hand and feet, and partial syndactyly of hands and feet were observed. A normal 46 XY cariotype, and a normal neural development were found.
Discussion. We present a case of Pfeiffer syndrome type 2 without visceral complications and normal neurologic development.
KEY WORDS Pfeiffer syndrome, neurological development.
Introducción
El síndrome de Pfeiffer se caracteriza clínicamente por braquicefalia con cráneo-sinostosis de la coronal, frente alta, hipertelorismo ocular, falange distal ancha del pulgar y del primer dedo del pie, sindactilia parcial de segundo y tercer dedo de manos, y segundo, tercero y cuarto dedos de los pies. Se hereda de forma autosómica dominante. 1 Cohen 2 en 1993 dio a conocer tres tipos de esta entidad, señalando que el tipo 2 se caracteriza por cráneo en trébol, proptosis ocular severa, daño severo del sistema nervioso central, sinostosis de articulación de codos, con muerte temprana. Desde 1964 a la fecha se han reportado cerca de 30 casos.
Este trabajo tiene como objetivo presentar un caso clínico de síndrome de Pfeiffer, en un niño de cinco años de edad, con cráneo en trébol, proptosis ocular severa, y aparentemente sin retardo mental.
Presentación del caso clínico
Paciente del sexo masculino de cinco años de vida, originario y residente de Tacotalpa, Tabasco, México; producto de la segunda gesta, padres de 19 años de edad al momento de nacer el propositus, no consanguíneos, sin antecedentes teratogénicos ni de padecimiento similar en otros miembros de la familia. Desarrollo del embarazo normal, de término, parto terminado en cesárea por desproporción cefalopélvica. Período neonatal sin complicaciones, desconociéndose somatometría al nacimiento. Sostuvo la cabeza a los cuatro meses de vida, bipedestación a los nueve meses y deambulación a los 13 meses.
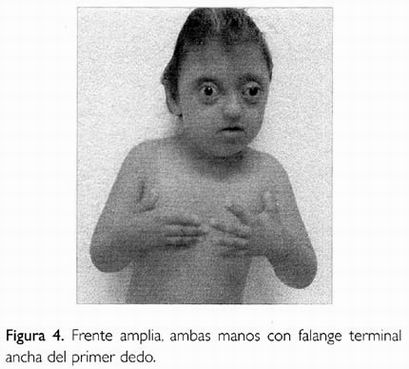
A la exploración física: peso 19 500 kg, talla 108 cm, circunferencia cefálica 52 cm. Edad aparente mayor a la real, íntegro, complexión delgada, cráneo en trébol, frente amplia y prominente (Fig. 1). Proptosis ocular, con antecedente de salida del globo ocular derecho en dos ocasiones, aplanamiento medio facial, puente nasal aplanado, paladar alto, pabellones auriculares de implantación baja y configuraciones en cruz del helix horizontal (Figs. 2 y 3). Cuello corto, primer dedo de manos y pies anchos de su falange distal, sindactilia parcial en manos y pies (Figs. 4 y 5). Dentro de los estudios de rutina la biometría hemática mostró: hemoglobina de 9 g/dL, química sanguínea y examen general de orina normales. Determinaciones de mucopolisacáridos totales y aminoácidos en orina de 24 horas resultaron negativos. El cariotipo en linfocitos de sangre periférica mostró un complemento cromosómico normal 46, XY. Radiológicamente se observó cráneo en trébol, con múltiples impresiones digitales (Fig. 6), aumento de grosor de última falange de primer dedo de manos y pies, sindactilia de segundo a cuarto dedos en ambos pies.





Discusión
Las acrocefalosindactilias son síndromes caracterizados por anormalidades del cráneo (cráneo-sinostosis), la cara (hipertelorismo, retromaxilismo), manos y pies (sindactilia ósea o cutánea). Se heredan de manera autosómica dominante, aunque también se han descrito casos espontáneos. Este grupo de entidades incluye síndromes severos con expresividad variable y distintos grados de penetrancia. 3
El síndrome de Pfeiffer es una acrocefalosindactilia genéticamente heterogénea. Tres subtipos clínicos han sido descritos basados en la severidad de la acrocefalosindactilia y de las manifestaciones asociadas 2 (Cuadro 1)
Muenke y col. 4 mostraron evidencias de mutación génica en el receptor 1 de crecimiento fibroblástico (FGR1) mapeado en el cromosoma 8p11.22-p12. Otros casos son causados por mutaciones en el receptor-2 de crecimiento fibroblástico (FGFR-2) que se encuentra en el cromosoma 10q25-q26 lo que sugiere que el síndrome de Pfeiffer es una entidad con heterogeneidad, correspondiendo esta mutación a la familia original reportada por Pfeiffer 5 en 1964, en la cual había ocho afectados en tres generaciones, con transmisión de hombre a hombre, y cuyas manifestaciones clínicas fundamentales fueron la falange terminal del primer dedo de manos y pies cortas y anchas. El síndrome de Pfeiffer es considerado como una acrocefalosindactilia, debiendo hacerse diagnóstico diferencial con el síndrome de Apert, el síndrome de Noack, y el tipo II del síndrome de Carpenter. 6-11 Cohen 2 en 1993 mencionó que el tipo 2 del síndrome de Pfeiffer cursa con cráneo en trébol, proptosis ocular severa, daño severo del sistema nervioso central, sinostosis de codos, primer dedo de manos y pies anchos, anomalías viscerales, y muerte temprana. El caso presentado aquí corresponde clínica y radiológicamente a un síndrome de Pfeiffer tipo 2, de cinco años de vida, sin complicaciones viscerales, y con desarrollo neurológico de acuerdo a su edad cronológica (Cuadro 2). Todos los casos reportados del síndrome de Pfeiffer tipo 2 y 3 han sido esporádicos.
REFERENCIAS
1. Teebi AS, Kennedy S, Chun K, Ray PN. Severe and mild phenotypes in Pfeiffer syndrome with aplice acceptor mutations in exon IIIc of FGFR2. Am J Med Genet 2002; 107: 43-7. [ Links ]
2. Cohen MC. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet 1993; 45: 300-5. [ Links ]
3. Freihofer HP. Syndrome 2. Pfeiffer syndrome. Ned Tijdschr Tandheelkd 1998; 105: 245-6. [ Links ]
4. Muenke M, Schell V, Hehr A. A common mutation in the fibroblast growth factor receptor I gene in pfeiffer syndrome. Nat Genet 1964; 90: 301-20. [ Links ]
5. Pfeiffer RA. Dominant erbliche akrocephalosyndaktylie. Z Kindarheilk 1964; 90: 301-20. [ Links ]
6. Rutland P. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nat Genet 1995; 9: 173-8. [ Links ]
7. Mckusick VA. Acrocephalosyndactyly type V. En: Mendelian inheritance in man. 12 a ed. Vol. 2. Philadelphia: The Johns Hopkins University Press; 1998. p. 26-7. [ Links ]
8. Shotelersuk V, Ittiwut C, Srivuthana S, Mahatumarat C, Lerdlum S, Wacharasindhu S. Distinct craniofacial-skeletal-dermatological dysplasia in a patient with W290C mutation in FGFR2. Am J Med Genet 2002; 113: 4-8. [ Links ]
9. Chun K, Teebi AS, Jung JH, Kennedy S. Genetic analysis of patients with the Saethre-Chotzen phenotype. Am J Med Genet 2002; 110: 136-43. [ Links ]
10. Rebelo N, Duarte R, Costa MJ, Leal MJ. Acrocephalosyndactyly the coalesced hand. Eur J Pediatr Surg 2002; 12: 49-55. [ Links ]
11. Hermanns P, Lee B. Transcriptional dysregulation in skeletal malformation syndrome. Am J Med Genet 2001; 106: 258-71. [ Links ]

