











 nueva página del texto (beta)
nueva página del texto (beta) Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Se define como muerte súbita (MS) a la interrupción brusca de todas las funciones vitales del cuerpo, manifestándose por pérdida permanente de las funciones cerebral, respiratoria y cardiovascular1. El término de muerte súbita cardiaca (MSC) se reserva para la muerte que acontece de forma rápida por colapso cardiovascular dentro de la primera hora de iniciados los síntomas premonitorios, generalmente causada por anormalidades cardiovasculares2. Cuando la MS ocurre en un niño menor de un año de edad, aparentemente sano, sin explicación después de una investigación exhaustiva se denomina muerte súbita infantil (MSI)3.
La MS es un evento trágico e inesperado que impacta seriamente a la población general y médica; cuando ocurre en un niño, es devastador para la familia y la comunidad. En México, no existe un registro nacional de MSC en la edad pediátrica. En 2015, el Instituto Nacional de Estadística y Geografía (INEGI) reportó 3,842 muertes por causas desconocidas o mal definidas (R95-R99), de las cuales, 514 correspondieron a MSI y 59 a muerte instantánea4. En los Estados Unidos, la MS de recién nacidos, niños y adolescentes tiene una incidencia de 1.3 a 8.5 por 100,000 pacientes/año5 y el centro para el control y prevención de enfermedades de los Estados Unidos (CDC) estima que aproximadamente 2,000 personas menores de 25 años morirán de manera súbita cada año en dicho país6.
Aunque la MSC, puede ocurrir en niños y jóvenes aparentemente sanos como el evento centinela en realidad, con mayor frecuencia esta es precedida por la presencia de signos y síntomas que pueden pasar inadvertidos o no son adecuadamente estudiados y que son secundarios a una enfermedad cardiovascular congénita o adquirida no sospechada.
Las enfermedades que predisponen a la MSC en niños pueden clasificarse en dos grandes grupos: a) enfermedades estructurales y b) enfermedades eléctricas.
Las enfermedades estructurales que predisponen a la MSC son aquellas en las que existe una anormalidad anatómica y por lo general aunque no siempre, pueden diagnosticarse con un ecocardiograma. Se trata de enfermedades en las que existe una malformación congénita del corazón que pueden ocasionar arritmias malignas como consecuencia de los cambios hemodinámicos propios de la cardiopatía o relacionados con la corrección quirúrgica; como por ejemplo la tetralogía de Fallot con riesgo de fibrilación ventricular (FV) postinsicional o las cardiopatías de ventrículo único con riesgo de MS por disfunción sinusal posterior a la cirugía de Fontan, entre otras. Otro grupo de enfermedades estructurales con riesgo de MSC son aquellas en las que en ausencia de una malformación congénita cardiaca, existen anormalidades en la estructura del corazón y/o de la vasculatura cardiaca. Dentro de este último grupo, se encuentran algunas enfermedades genéticas con afección vascular como el síndrome de Marfan con aneurisma aórtico y otras enfermedades adquiridas como la enfermedad de Kawasaki, la miocarditis y las miocardiopatías en sus diferentes presentaciones: miocardiopatía dilatada (MCD), miocardiopatía hipertrófica (MCH), miocardiopatía arritmiógena, miocardiopatía restrictiva (MCR) y la miocardiopatía no compactada (MCNC).
Por otro lado, las enfermedades eléctricas que pueden ocasionar MSC, constituyen anormalidades del ritmo eléctrico cardiaco que se presentan en el contexto de un corazón estructuralmente normal. En este grupo, destacan de manera especial las enfermedades arritmogénicas hereditarias, también denominadas canalopatías. El sustrato genético de las enfermedades arritmogénicas hereditarias radica en mutaciones en genes que codifican diferentes canales iónicos presentes en la membrana plasmática de los cardiomiocitos o en genes que forman parte de las proteínas auxiliares de dichos canales7,8 (fig. 1). Estas enfermedades comparten dos características fundamentales: su carácter hereditario y el riesgo de desencadenar arritmias malignas del tipo de la taquicardia ventricular (TV), que puede degenerar en FV y por este mecanismo ocasionar síncope, convulsiones y/o MSC. Otras enfermedades arritmogénicas que pueden causar MSC como el síndrome de Wolff-Parkinson-White, disfunción sinusal congénita, bloqueo aurículo-ventricular completo entre otras, se diferencian del primer grupo en que el riesgo de MSC por arritmias malignas es más bajo. Aunque también puede existir un componente genético bien definido.
El objetivo del estudio fue conocer la prevalencia y el espectro de las enfermedades que predisponen a la MSC en niños a partir de una muestra obtenida en el Hospital Infantil de México Federico Gómez (HIMFG), así como señalar los principales signos y síntomas que estuvieron presentes antes del diagnóstico, para favorecer la sospecha de estas enfermedades por el personal de salud, la referencia oportuna a un hospital de tercer nivel y con ello la disminución de la incidencia de MSC en la edad pediátrica.

Métodos
Diseño
Se realizó un estudio observacional, analítico, longitudinal y ambispectivo en el Laboratorio de Investigación en Cardiopatías Congénitas y Arritmias del HIMFG. La inclusión de los pacientes se realizó a partir del 1 de enero de 2014 hasta el 31 de julio de 2015. El periodo de seguimiento incluye desde la fecha en que se estableció el diagnóstico hasta el 31 de diciembre de 2015. El protocolo de investigación fue aprobado por los comités de Investigación, Ética y Bioseguridad del HIMFG. Todos los pacientes y sus familiares incluidos aceptaron la participación en el estudio y firmaron cartas de consentimiento informado.
Población de estudio
Se incluyeron dos grupos de pacientes: Grupo 1. Niños con miocardiopatías.
Grupo 2. Niños con enfermedades arritmogénicas hereditarias (canalopatías).
Para fines de este proyecto de investigación, no se incluyeron otras enfermedades estructurales ni eléctricas que pueden ocasionar MSC como las cardiopatías congénitas complejas, los síndromes genéticos con afección vascular ni otras enfermedades arritmogénicas diferentes a las canalopatías.
Todos los pacientes se evaluaron clínicamente al momento de la inclusión al estudio. Los datos obtenidos de los expedientes hospitalarios, de la evaluación clínica y durante el periodo de seguimiento se concentraron en una base de datos diseñada especialmente para este proyecto de investigación. En todos los casos, estudiamos a todos los miembros de la familia disponibles para la búsqueda intencionada de las enfermedades que causan la MSC y para analizar los patrones de herencia mendeliana.
Evaluación clínica
A todos los pacientes se les realizó una evaluación cardiológica que incluyó la historia clínica personal y familiar, examen físico, evaluación de la clase funcional (CF) de acuerdo a la nueva clasificación basada en la edad propuesta por Ross9. Se interrogó de manera intencionada y exhaustiva sobre los primeros signos y síntomas identificados y su evolución en el tiempo, así como el antecedente de la muerte repentina en algún miembro de la familia.
Se realizaron los siguientes estudios cardiológicos básicos: electrocardiogramas de superficie de 12 derivaciones (ECG-12) y electrocardiogramas con derivaciones precordiales altas10, estos estudios fueron analizados por tres observadores independientes.
Ecocardiograma bidimensional (Eco-2d). De forma ciega en relación con los datos clínicos, familiares y eléctricos, alguno de los tres ecocardiografistas del departamento de cardiología realizó Eco-2d utilizando un ecocardiógrafo Phillips IE33. Siguiendo las recomendaciones de la Sociedad Americana de Ecocardiografía11, se obtuvieron utilizando el modo M, la fracción de eyección del ventrículo izquierdo (FEVI), el diámetro de la pared posterior del ventrículo izquierdo en diástole (PPVID) y el diámetro telediastólico del ventrículo izquierdo (DDVI) y utilizando el Doppler pulsado, el gradiente de obstrucción al tracto de salida del ventrículo izquierdo (VI). Basados en estudios previos, un índice PPVID/DDVI < 0.14 en pacientes con miocardiopatía dilatada (MCD) así como un comportamiento obstructivo al tracto de salida del VI se consideraron datos ecocardiográficos de alto riesgo de MSC en pacientes con MCD y MCH respectivamente12-14.
El monitoreo electrocardiográfico ambulatorio tipo Holter de 24 h se realizó utilizando un equipo Phillips Zymed Digitrak Plus y los resultados fueron analizados por una electrofisióloga pediatra.
Para la determinación de la CF, a los pacientes del grupo 1 y mayores de 9 años, se les realizó la determinación de los niveles séricos de la porción N-terminal del péptido natriurético tipo B (NT-pro-NPPB) y la prueba de esfuerzo cardiopulmonar utilizando un protocolo de rampa en banda sin fin limitada por síntomas, con análisis de gases respiratorios simultáneo. El consumo de oxígeno máximo (VO2) se definió como el VO2 máximo alcanzado durante el ejercicio, expresado en ml/min/kg. El porcentaje de predicción de VO2 en relación con la edad y el sexo del paciente se calculó según las guías15.
Se diagnosticó insuficiencia cardiaca (IC) refractaria cuando los pacientes se mantuvieron en CF III o IV a pesar de estar recibiendo dosis máximas del tratamiento óptimo para IC.
Los pacientes fueron evaluados mensualmente o antes si presentaron algún evento clínico.
Estudio genético
Los patrones de herencia mendeliana en las familias fueron analizados por una genetista e investigadora del HIMFG. Se registró si se había realizado algún estudio genético en el HIMFG o en otra institución y el resultado del mismo.
Tratamiento
El protocolo de tratamiento óptimo para IC en niños en el HIMFG incluye diuréticos, inhibidor de la enzima convertidor de angiotensina, þ-bloqueador y digital. Las dosis de los fármacos se ajustan de acuerdo al peso, la tolerancia y a la evolución clínica. Los pacientes en CF IV se hospitalizaron para la administración intravenosa de inotrópicos y diuréticos y para el apoyo ventilatorio en algunos casos. La terapia de resincronización cardiaca mediante el implante de un marcapasos tricameral endocárdico o epicárdico se llevó a cabo en los pacientes con IC refractaria. Un desfibrilador automático implantable (DAI) se implantó para la prevención primaria o secundaria de MSC en los casos de alto riesgo siempre que fue posible. Los pacientes recibieron un trasplante cardiaco (TC) siempre que se obtuvo la donación de un órgano compatible a su peso y grupo sanguíneo.
Análisis estadístico
Para las variables cuantitativas se utilizó estadística descriptiva: media, desviación estándar, valores mínimos, máximos y porcentajes. Para las variables nominales se utilizó estadística descriptiva con frecuencias, porcentajes y representación con gráficos de barras.
La tasa de incidencia se calculó utilizando la siguiente
fórmula:
Tasa de Incidencia =Número de casos nuevos / población total ∗ periodo de tiempo determindo
La prevalencia se calculó utilizando la siguiente fórmula:
Prevalencia = Número de casos existentes / población total
La prevalencia de periodo (PP) se calculó utilizando la siguiente fórmula:
Donde: C0 = total de casos existentes al inicio del estudio (tiempo 0, t0).
I = casos nuevos que ocurren durante el periodo de estudio.
N = total de la población al inicio del estudio.
Para el análisis de la concordancia interobservador, calculamos el coeficiente kappa utilizando la siguiente fórmula:
Se realizó la valoración del coeficiente kappa de acuerdo a la escala de Landis y Koch16.
Resultados
Se incluyeron cincuenta y nueve niños de 0 a 17.66 años (8 ± 5 años), 33 mujeres (55.9%), con un periodo de seguimiento de 5.4 ± 0.2 años. Veintidós (37.58%) fueron referidos por primera vez, en diecinueve el diagnóstico correcto de la enfermedad se había establecido en el hospital de referencia (32.2%).
La mayoría de los pacientes provenían del Estado de México y de la Ciudad de México (tabla 1).
Prevalencia de las enfermedades que predisponen a la muerte súbita cardiaca en niños
De acuerdo a los registros en el Departamento de Archivo Clínico y Estadística del HIMFG, hasta diciembre de 2014, existían 6,187 pacientes vivos con registro hospitalario activo. Durante el año de 2014, se otorgaron 4,785 consultas de primera vez. Con esta información, utilizando las fórmulas previamente descritas y considerando que del 1 de enero de 2014 al 31 de julio de 2015 se identificaron 59 niños con alguna de las enfermedades que predisponen a la MSC en nuestro hospital, se calculó una tasa de incidencia de estas enfermedades en el HIMFG de 5.4/1,000 pacientes/año; con una prevalencia de 5.3/1,000 pacientes/año y considerando que durante el periodo de este estudio se incluyeron 26 nuevos casos, la prevalencia de periodo (PP) es de 9.5/1,000 pacientes/año.
Tabla 1 Distribución por estados de origen de los niños con enfermedades con riesgo de muerte súbita cardiaca del Hospital Infantil de México Federico Gómez
| Estado | Frecuencia | Porcentaje % |
| Baja California Sur | 2 | 3.40 |
| Chiapas | 3 | 5.10 |
| Estado de México | 16 | 27.10 |
| Guanajuato | 2 | 3.40 |
| Guerrero | 3 | 5.10 |
| Hidalgo | 2 | 3.10 |
| Jalisco | 1 | 1.70 |
| Michoacán | 2 | 3.40 |
| Ciudad de México | 12 | 20.30 |
| Nuevo León | 1 | 1.70 |
| Oaxaca | 1 | 1.70 |
| Puebla | 6 | 10.20 |
| San Luis Potosí | 1 | 1.70 |
| Tabasco | 2 | 3.40 |
| Veracruz | 4 | 6.80 |
| Zacatecas | 1 | 1.70 |
| Total | 59 | 100 |
Espectro de las enfermedades que predisponen a la muerte súbita cardiaca en niños
Cuarenta niños se diagnosticaron con una miocardiopatía (67.8%) correspondiendo a MCD 25 casos (62.5%), MCH 9 (22.5%), MCR 4 (10%) y MCNC 2 (5%). En 19 niños, se estableció el diagnóstico de alguna enfermedad arritmogénica hereditaria (32.2%), síndrome de QT largo (SQTL) en 17 (89.48%), enfermedad de repolarización precoz (ERS) en 1 (5.26%) y síndrome de QT corto (SQTC) en 1 (5.26%) (fig. 2). Se observó una diferencia estadísticamente significativa en el tiempo de seguimiento entre los niños con miocardiopatías (5.88 ± 0.91 años) y los que tuvieron una enfermedad arritmogénica hereditaria (2.30 ± 0.35 años) (p >0.05); ya que todos los 19 pacientes del segundo grupo fueron diagnosticados a partir de marzo de 2012.
Primeros signos y síntomas de la enfermedad
Los primeros signos y síntomas aparecieron desde el nacimiento y hasta los 15 años (5.12 ± 0.75 años), en 20 antes de los 12 meses (35.59%). El diagnóstico se estableció desde el nacimiento y hasta los 17 años (6.4± 0.82 años). El tiempo, entre la aparición de los primeros signos o síntomas y el diagnóstico de la enfermedad fue de 0-13.3 años (1.2 ± 2.5 años).
De manera general, los primeros signos y síntomas más frecuentes fueron: disnea en 25 (43%), fatiga en 25 (43%) y palpitaciones en 12 (21%). En el grupo 1, la disnea fue el primer síntoma en 24 pacientes (60%) y en el grupo 2 el síncope en 7 (39%), seguido de palpitaciones en 5 (26%) (fig. 3). En 4 pacientes (22%), la MS abortada (MSA) fue el evento centinela de la enfermedad sin identificarse signos y síntomas previos.

Figura 2: Prevalencia de las enfermedades con riesgo de muerte súbita cardiaca en niños del Hospital Infantil de México Federico Gómez. Se muestra el número de casos diagnosticados para cada enfermedad. ERS: síndrome de repolarización temprana; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MCNC: miocardiopatía no compactada; MCR: miocardiopatía restrictiva; SQTC: síndrome de QT corto; SQTL: síndrome de QT largo
Grupo 1. Pacientes con miocardiopatías
Evaluación clínica
Después de optimizar el tratamiento para IC, que incluyó a la TRC en algunos casos, once pacientes se encontraban en CF I (31.4%), 12 en CF II (34.3%), 7 en CF III (20%) y 5 en CF IV (14.3%).
Evaluación ecocardiográfica
La FEVI inicial fue de 14 --78% (44 ± 26%). Once pacientes con MCD tuvieron un PPVID/DDVI <0.14 y en 2 pacientes con MCH se observó un patrón obstructivo.
Análisis del electrocardiograma de superficie de 12 derivaciones
Todos los pacientes con miocardiopatías mostraron una desviación anormal del eje eléctrico del corazón a la izquierda y voltajes elevados del complejo QRS. En veintisiete pacientes (67.5%) se observaron trastornos de la repolarización ventricular con anormalidades en la morfología de la onda T en 24 (88%), en el segmento ST en 9 (33%) y un intervalo QT corregido (QTc) prolongado en 22 (55%). El QTc fue aún más prolongado en el grupo de MCD (359-607 mseg; m: 481 ± 75 mseg) donde 15/25 (60%) tuvieron un QTc mayor de 460 mseg (coeficiente kappa de 0.88 - 0.96).

Figura 3: Prevalenciade signos y síntomas en los niños antes del diagnóstico de las enfermedades con riesgo de muerte súbita cardiaca.
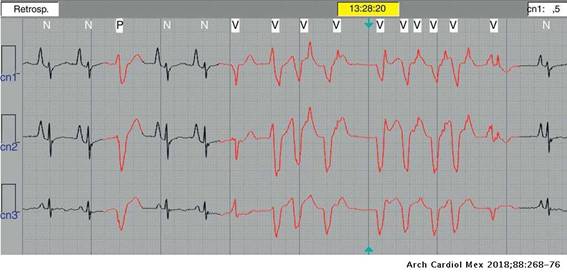
Monitoreo electrocardiográfico tipo Holter de 24 h
Veinte pacientes (50%) mostraron alguna anormalidad durante el Holter de 24 h. Taquicardia sinusal persistente en 12 (63.2%), extrasístoles auriculares frecuentes de presentación sencilla en seis (31.5%), extrasístoles ventriculares poco frecuentes en uno (5.3%) y en otro paciente (5.3%) se documentó un evento de TV monomórfica no sostenida de 9 latidos (fig. 4).
Tratamiento
Todos los niños con MCD recibieron el tratamiento óptimo establecido en el HIMFG para IC en niños. Diez con MCD (25%) recibieron TRC; por vía epicárdica 3 (30%) y endocárdica 7 (70%); todos en IC refractaria. Veintiuno (52.5%), ingresaron a espera de TC, ocho se trasplantaron (38.09%). El tratamiento de los niños con los otros tipos de miocardiopatías fue variable y dependió de la condición clínica y ecocardiográfica. Una niña con MCH y dolor precordial asociado a isquemia subepicárdica recibió isosorbide (5 mg/día), se trasplantó durante el periodo de estudio y se mantiene estable. Un niño con MCH falleció 72 h después de la miomectomía quirúrgica. Dos niños con MCH obstructiva (22%) recibieron un DAI y continúan en vigilancia, en CF I.
Grupo 2. Pacientes con enfermedades arritmogénicas hereditarias
Diecinueve pacientes (32.2%) tuvieron una enfermedad arritmogénica hereditaria, diecisiete SQTL (89.48%), uno ERP (5.26%) y uno SQTc (5.26%). Todos los pacientes con SQTL recibieron la dosis máxima tolerada de þ-bloqueador. El þ- bloqueador utilizado en todos los casos fue propranolol. Tres pacientes con SQTL (17.64%) recibieron un DAI; dos por MSA (prevención secundaria) y uno con eventos frecuentes de TV sin síncope (prevención primaria). El QTc en estos pacientes fue de 503 ± 81 (471-624) mseg.
Antecedente de muerte súbita cardiaca
En seis pacientes, existió el antecedente familiar de MS (10%) y cuatro tuvieron un evento de MSA (6%). Todos los pacientes con MSA tuvieron diagnóstico de SQTL, en todos la MSA fue el evento centinela de la enfermedad, dos de ellos menores de un año (MSI).
Estudio genético y herencia mendeliana en las familias Ningún paciente o familia había sido previamente estudiado genéticamente en otra institución.
La evaluación familiar identificó familiares afectados de primer y segundo grado en 9 pacientes (15.25%) con lo que se reconocieron los siguientes patrones de herencia mendeliana: en tres pacientes con SQTL, el patrón de herencia autosómico dominante se determinó debido a que al caso índice lo antecedía un familiar afectado en primer grado, además de observar familiares (mujeres y hombres) afectados en más de dos generaciones y herencia varón- varón. En dos con MCD los patrones de herencia encontrados fue herencia autosómica dominante en una niña con afectados en tres generaciones y herencia varón-varón y en otra herencia autosómica recesiva sin antecedente de consanguinidad paterna. Además, identificamos dos familias afectadas con MCH familiar. En la primera, al caso índice le antecede su padre y dos hermanos afectados con la enfermedad; por lo que existe herencia autosómica dominante y herencia varón-varón. La otra familia presenta antecedente familiar de varios afectados en la misma hermandad y sin antecedente de consanguinidad por parte de los padres, sin embargo, reconocemos la herencia autosómica recesiva en la familia. Un caso índice con MCR presenta herencia autosómica dominante, debido a que en su genealogía existen varios miembros afectados (mujeres y hombres) en tres generaciones. Por último, un caso índice con ERS presenta antecedente materno para la misma enfermedad y un hermano con SQTC, como expresividad variable de enfermedades que predisponen a la MSC con herencia autosómica dominante.
Mortalidad
Ocho pacientes fallecieron (13.55%); siete con MCD (11.86%) y uno con MCH (1.69%). La causa de muerte en MCD fue IC refractaria en 3 y MSC en 3; los seis se encontraban en espera para TC. Un niño con MCD falleció por probable sepsis nueve meses después de TC. El paciente con MCH falleció 72 horas posteriores a la cirugía de miomectomía septal. Ninguno de los pacientes con enfermedades arritmogénicas hereditarias falleció durante el periodo de estudio.
Discusión
En México, las enfermedades que predisponen a la MSC en la edad pediátrica son poco conocidas por la comunidad general y médica. El registro de muerte en la edad pediátrica asociada a «causas desconocidas» apoya la posibilidad de que muchos niños fallecen de MSC sin haberse sospechado la presencia de la enfermedad cardiaca. Con base en esta preocupación se diseñó este estudio de investigación.
Aunque la prevalencia presentada es la obtenida de una muestra del HIMFG, nos brinda una idea del número de pacientes con estas enfermedades y su espectro, atendidos al menos en uno de los hospitales pediátricos más grandes de nuestro país. La incidencia y prevalencias obtenidas se calcularon con base en la población de pacientes activos y en el número de pacientes de primera vez que acuden anualmente. Una prevalencia estimada en el periodo de estudio de 9.5/1,000/año debe ayudar a los médicos generales, pediatras y cardiólogos a reconocer que estas enfermedades no son tan raras como se piensa. Aunque las enfermedades estructurales ocuparon dos terceras partes del total de los pacientes, las enfermedades arritmogénicas hereditarias se reconocieron todas a partir de 2012 después de iniciar un programa para la búsqueda intensiva de las mismas en nuestro hospital y después de la capacitación a médicos especialistas en formación en el cálculo del QTc y en la búsqueda de otros parámetros eléctricos anormales en el ECG-12, así que la prevalencia de periodo para este grupo es aún mayor.
En México contamos con cifras aproximadas de MSC basadas en datos reportados por INEGI, pero muchos casos de MSC no son registrados correctamente por lo que no estamos en posibilidad de establecer comparaciones válidas con los registros de otros países. En 2013, Canadá tenía una incidencia de MSC en niños de 15-19 años de 1.01 por cada 100,000 personas17, en Asia la MSC ocurre en 40 casos por cada 100,000 personas18, en Irlanda el registro nacional de MSI, incluye 519 casos en un periodo de 15 años (1995-2009)19.
Cuando se analiza por separado el grupo de personas jóvenes, la principal enfermedad estructural a nivel mundial que ocasiona MSC, es la MCH20. Por otro lado, en el grupo de niños, el registro de miocardiopatías pediátricas de Estados Unidos estima una incidencia de MSC de 0.47 por cada 100,000 nin˜os con MCH y 0.58 por cada 100 000 niños con MCD21.
Una meta urgente en términos de salud en la edad pediátrica, es el buscar estrategias para reducir de manera significativa la MSC en los niños. Es necesario establecer políticas de prevención primaria y secundaria.
Para realizar prevención primaria, se debe difundir el conocimiento sobre las enfermedades que predisponen a los niños a este riesgo.
La sospecha de las enfermedades estructurales que causan la MSC se establece más tempranamente que en el grupo de niños con enfermedades arritmogénicas ante la presencia de una exploración física sugestiva de una alteración anatómica, la cual luego se corrobora con el ecocardiograma.
En el grupo de niños con miocardiopatía, el riesgo de MSC debe evaluarse al momento del diagnóstico y durante el seguimiento. La MSC puede aparecer como resultado de arritmias malignas. Frecuentemente, se trata de niños en etapas avanzadas de la enfermedad. En estos casos, además de la optimización del tratamiento de la IC se debe evaluar si el paciente requiere de tratamiento antiarrítmico, así como analizar si otras opciones terapéuticas como la miomectomía o la ablación con alcohol en casos seleccionados de MCH o un TC por IC refractaria en todos los tipos de miocardiopatías deben ofrecerse. El implante de un DAI está indicado en los sobrevivientes de un paro cardiaco, después de un evento de TV sintomática y en casos de síncope recurrente cuando se han eliminado otras causas reversibles22. Además del ECG-12, un estudio Holter de 24 h brinda información relevante como sucedió con uno de nuestros casos en quien se identificó un evento de TV no previamente documentada. En casos más complejos puede ser necesario un estudio electrofisiológico para la toma de decisiones.
Las enfermedades arritmogénicas hereditarias representan un mayor reto diagnóstico, debido a que estas se aparecen en presencia de un corazón estructuralmente normal en un niño o adolescente aparentemente sano. La piedra angular para el diagnóstico es el ECG-12, que con frecuencia muestra anormalidades que permiten sospechar la mayoría de estas enfermedades. Sin embargo, aunque su costo es considerablemente menor que el de un ecocardiograma, el ECG-12 se solicita en muy pocos casos y en otros no es adecuadamente interpretado.
Si bien la MSC puede presentarse como el evento centinela de estas enfermedades, habitualmente estos niños presentan signos y síntomas que suelen pasar inadvertidos. Los niños incluidos en este estudio presentaron disnea, síncope, palpitaciones y dolor precordial al menos un año antes de la referencia, pero en un caso los síntomas estuvieron presentes durante 13 años antes de establecerse el diagnóstico por lo que la realización de un ECG-12 y un examen físico exhaustivo en todo niño que acude a consulta pediátrica o cardiológica con cualquier signo o síntoma posiblemente cardiaco está indicado. Por otro lado, conocer los valores electrocardiográficos de referencia de normalidad en niños deberá ser una meta de salud general y el ser capaces de identificar patrones eléctricos peligrosos debería ser requisito en los médicos generales que brindan el primer nivel de atención.
Aunque la incidencia de MSI ha ido disminuyendo desde la campaña de regreso al sueño en posición supina, se ha estabilizado en alrededor de 0,5-0,6 por 1,000 nacidos vivos en los Estados Unidos23. Después de evaluar a 34,442 recién nacidos, Schwartz informó una asociación significativa con una prolongación del intervalo QTc y MSI24. El QTc medido en el tercer o cuarto día de vida, de los lactantes que murieron súbitamente, fue estadísticamente más prolongado que el de los lactantes que sobrevivieron hasta el año de edad (435 ± 45 mseg vs. 400 ± 20 mseg, p < 0,01). Debido a esto, Schwartz plantea que la exploración neonatal de rutina por ECG y la profilaxis con betabloqueantes pueden estar justificadas. Basados en estas y en otras observaciones, nosotros recomendamos la realización de un ECG-12 en todo recién nacido como parte de la rutina de evaluación cardiológica del recién nacido y de manera obligada en los recién nacidos con algún antecedente familiar importante. Para soportar localmente esta recomendación, en el HIMFG hemos iniciado un proyecto piloto nacional y multicéntrico de tamizaje electrocardiográfico en el recién nacido.
El ecocardiograma fetal es de gran utilidad para el diagnóstico temprano de algunas cardiopatías congénitas y también permite la identificación de arritmias fetales por medio de mediciones con doppler25 lo que favorece la identificación de enfermedades que predisponen a MSC aún antes del nacimiento.
En términos de prevención secundaria, se requieren programas de reanimación cardiopulmonar básica y de manejo del desfibrilador externo automático (DEA) para la población general. Estos programas podrían iniciarse en las escuelas y dirigirse a los jóvenes en los niveles de educación media y media superior, así como una capacitación a los padres, cuidadores y maestros, además de la necesidad de contar con programas de «territorios cardioprotegidos» que garanticen la existencia de un DEA en lugares con concentraciones altas de niños y jóvenes.
En el HIMFG estamos realizando la búsqueda de mutaciones en genes asociados por medio de la secuenciación masiva de nueva generación lo que nos permitirá describir la base genética de estas enfermedades en niños mexicanos, establecer asociaciones fenotipo-genotipo, patrones de segregación y sentar las bases para la realización de estudios funcionales que nos ayuden a identificar marcadores de riesgo y poder realizar una cardiología preventiva más eficiente.
Conclusiones
Es posible prevenir la MSC en niños si las enfermedades que les confieren este riesgo se diagnostican oportunamente. La prevalencia de periodo de las enfermedades que predisponen a la MSC en niños en el HIMFG fue considerablemente elevada de 9.5/1,000 pacientes/año. La presencia de palpitaciones, disnea, dolor precordial y síncope entre otros signos y síntomas sutiles en niños, deben analizarse cuidadosamente con una evaluación cardiológica completa que incluya ECG-12. Todo niño con sospecha de alguna de estas enfermedades, debe referirse a un hospital de tercer nivel para su estudio exhaustivo. Una vez establecido el diagnóstico, el estudio integral en todos los miembros disponibles de la familia está indicado.

