










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.82 no.4 Ciudad de México oct./dic. 2012
https://doi.org/10.1016/j.acmx.2012.06.006
Artículo de opinión
¿Es la hipertensión sensible a sal una enfermedad inflamatoria? Papel de los linfocitos y macrófagos
Is salt sensitive hypertension an inflammatory disease? Role of lymphocytes and macrophages
Rafael González-Toledo y Martha Franco
Departamento de Nefrologia y Fisiología Renal, Instituto Nacional de Cardiología Ignacio Chávez, México D.F., México.
Autor para correspondencia:
Rafael González-Toledo.
Instituto Nacional de Cardiología Ignacio Chávez.
Juan Badiano 1, Delegación Tlalpan, C.P. 14080, México D.F., México.
Teléfono: +55732911.
Correo electrónico: rtoledo29@hotmail.com
Recibido el 2 de marzo de 2012
Aceptado el 8 de junio de 2012
Resumen
La hipertensión arterial sistémica es un problema de salud pública. Esta entidad afecta al 43% de la población mexicana y es considerada una de los principales factores de riesgo para el desarrollo de eventos vasculares cerebrales, insuficiencia cardiaca e insuficiencia renal. La prevalencia de hipertensión arterial sistémica se ha incrementado durante las últimas décadas debido a la adopción de una dieta alta en sal. Existe evidencia de que la hipertensión sensible a sal se acompaña de alteraciones estructurales renales como dilatación tubular, fibrosis intersticial en parches, expresión de osteopontina e infiltrado túbulo intersticial de linfocitos y macrófagos, que impiden una excreción urinaria de sodio adecuada y en consecuencia, favorecen el desarrollo de HAS. De forma experimental se ha demostrado que estas alteraciones estructurales tienen una naturaleza inflamatoria y que la administración de medicamentos inmunosupresores disminuye la lesión tisular y mejoran el control de la presión arterial. En conjunto, los conocimientos derivados de los estudios de hipertensión sensible a sal pueden derivar en el desarrollo de nuevos fármacos destinados a mejorar el pronóstico asociado a la hipertensión arterial sistémica.
Palabras clave: Hipertensión; Cloruro de sodio; Inflamación; Linfocitos; Macrófagos; México.
Abstract
High blood pressure is a public health problem. This entity affects 43% of the mexican population and is considered a major risk factor for development of stroke, cardiac failure and chronic kidney disease. Hypertension prevalence has increased over the last decades, mainly because of high salt diet. There is evidence showing that salt-sensitive hypertension develops structural changes as tubular dilation, patchy interstitial fibrosis, osteopontin expression and lymphocytic/macrophage tubulointerstitial infiltrate that blunts urinary sodium excretion and therefore promotes HBP. It has been shown that this structural damage has an inflammatory origin and that immunosuppresant drugs down-regulates tissular injury and improves blood pressure control. In summary, this salt-sentitive hypertension data can be used in development of new and potent blood pressure drugs.
Keywords: Hypertension; Sodium chloride; Inflammation; Lymphocytes; Macrophages; Mexico.
Introducción
La hipertensión arterial sistémica (HAS) es un problema de salud pública que aumenta conforme se incrementa la edad de la población. Es el principal factor de riesgo para la presencia de eventos vasculares cerebrales, enfermedad cardiaca e insuficiencia renal.
En Estados Unidos en el año 2000, la Encuesta Nacional de Salud y Nutrición (NHANES, por sus siglas en inglés) reportó una prevalencia del 29%, es decir que cerca de 58.5 millones de individuos tenían cifras tensionales superiores a 140mmHg (presión sistólica) y/o 90mmHg (presión diastólica)1.
Las cifras en México son más alarmantes. La Encuesta Nacional de Salud y Nutrición (ENSANUT) reportó que en 2006 la prevalencia de HAS era de 43.1%2. El estado más afectado era Sonora, con 58.4% de prevalencia, mientras que el menos hipertenso era Morelos con 34.4%. Como puede observarse, incluso el estado con menos sujetos afectados tiene una media superior a la reportada en las estadísticas internacionales. Otro aspecto que es importante mencionar es que del total de los pacientes hipertensos, sólo el 40% conoce el diagnóstico, el 30% de éstos recibe tratamiento y únicamente el 15% del total de individuos tratados están bien controlados2.
Como lo ilustra la figura 1, durante las últimas décadas se ha observado un aumento en la prevalencia de HAS3. Hay evidencia de que ese incremento se asocia con la introducción y adopción del estilo de vida y dieta occidentales (rica en grasas saturadas, carbohidratos refinados y alto contenido de sal)3. Hasta años recientes había discusión en cuanto al efecto que tiene la carga de sodio (Na) sobre el control de la tensión arterial (TA), debido a que no todos los sujetos muestran la misma respuesta al ser sometidos a dieta alta en cloruro de sodio (NaCl).
El grupo de Weinberg4 fue de los primeros en establecer que la respuesta al Na es diferente en sujetos jóvenes y adultos/adultos-mayores. Mediante la exposición de los pacientes a una sobrecarga de Na y posteriormente a una depleción del mismo, establecieron que los individuos mayores de 60 años tenían una mayor sensibilidad de la TA al Na que los pacientes más jóvenes. La TA se modificaba más de 10 mmHg al comparar las cifras obtenidas tras la administración de solución salina, con aquellas encontradas después de la depleción de sal4. Además de los resultados anteriores, Midgley en un meta-análisis5 demostró que los estudios clínicos en los que se evaluaba el efecto de la depleción del Na dietético sobre las cifras de TA observaban resultados diferentes en sujetos jóvenes comparados con aquellos encontrados en adultos mayores. En promedio, por cada 100 mmol de depleción en la ingesta diaria de Na, se encontraba una disminución de 6.3 mmHg en la TA sistólica de los pacientes mayores de 60 años y de 2.4 mmHg en los pacientes más jóvenes; de manera similar, la disminución en la TA diastólica fue de 2.2 mmHg y 0.1 mmHg, respectivamente5. La HAS que se presenta por la ingesta de sal necesariamente requiere de la participación del riñón6.
Definición
La hipertensión sensible a sal (HSS) es el incremento de la TA media mayor a 10mmHg después de ingerir una dieta alta en Na, en un contexto de depleción de sal inducido por diurético y/o dieta hiposódica7.
El contenido de Na extracelular determina el volumen de agua corporal total, y por ende, el volumen plasmático1. Las modificaciones en el control de Na del organismo son muy importantes porque llevan a alteraciones de la TA. La incapacidad para excretar este catión a través de la orina invariablemente se asocia a HAS7. Una de las principales funciones del riñón es la regulación del volumen extracelular mediante la excreción de Na y agua, y se lleva a cabo mediante 2 mecanismos: 1) el sistema renina-angiotensina-aldosterona (SRAA), y 2) la excreción de Na urinario (NaU) secundaria a los cambios en la perfusión renal, mecanismo conocido como natriuresis de presión (a mayor presión en la arteria renal, mayor excreción de NaU)8.
Natriuresis de presión
El término ''natriuresis de presión'' se refiere al mecanismo por el cual los riñones controlan la TA mediante la modificación del volumen corporal. En condiciones de balance, cuando la TA aumenta, los riñones incrementan la excreción de Na y agua, disminuyendo el volumen plasmático y ocasionando que la presión regrese a valores basales.
La importancia de la natriuresis de presión en el control de la TA se puso en evidencia mediante estudios realizados por Guyton9: administró el 30% del volumen sanguíneo total a perros y observó que el incremento del gasto cardiaco y la TA, ocasionaban también aumento del gasto urinario y de la excreción de NaU, normalizándose las cifras de TA a las 2 horas de haber administrado la carga de volumen.
En la figura 2 se ilustran los mecanismos que controlan la TA después de una modificación súbita. Como puede observarse, los mecanismos de control nervioso (barorreceptores, quimiorreceptores y sistema nervioso central) son los primeros en actuar para estabilizar la TA. La excreción de NaU tiene un papel incipiente a los 60 minutos y preponderante, y prácticamente único después de las 8-16 horas de haberse presentado el cambio de TA10,11. El incremento de la TA aumenta también el flujo sanguíneo renal, que a su vez incrementa el flujo del líquido tubular que pasa a través de la mácula densa. En este punto se desencadena un mecanismo de retroalimentación entre mácula densa y arteriola aferente consistente en la vasoconstricción de esta última e incremento de la resistencia vascular, teniendo como efecto final la elevación de la resistencia renal total y disminución del flujo sanguíneo renal.
Además de los mecanismos anteriores, el aumento de flujo tubular a través de la mácula densa disminuye la secreción de renina y la formación de angiotensina II (AngII) (menor actividad del SRAA). Debido a que la AngII tiene un efecto vasoconstrictor sobre la arteriola eferente, la reducción en la concentración del péptido se acompaña de disminución en la resistencia arteriolar y de menor filtración glomerular (FG)11.
La excreción de NaU depende de la relación de 2 factores: la FG y la resorción tubular de Na. Como se ha mencionado anteriormente, cuando la TA aumenta se llevan a cabo mecanismos que mantienen estable o disminuyen la FG, por lo que la mayor eliminación de NaU depende de la menor resorción tubular12. Algunos estudios han encontrado que esta menor captación tubular de Na puede estar relacionada con aumento de la presión intersticial renal12,13, aumento del flujo sanguíneo medular renal14 e incremento de prostaglandinas y del sistema calicreína-cininas15; sin embargo, el estudio de estos mecanismos está fuera del objetivo del presente trabajo. En la figura 3 se observa un esquema que describe los mecanismos que participan en la natriuresis de presión.
La natriuresis de presión se representa gráficamente mediante una curva denominada curva de función renal (fig. 4). Esta figura registra en las abscisas la TA, y en las ordenadas la eliminación de NaU16. En esta gráfica se puede observar que conforme aumenta la TA, llega a un punto en el que la excreción de NaU aumenta exponencialmente, de tal forma que pequeños cambios de presión inducen grandes cambios en la excreción de NaU. Del mismo modo puede observarse que en condiciones de balance la ingesta de Na hasta7u8 veces el valor normal, no modifica la TA.
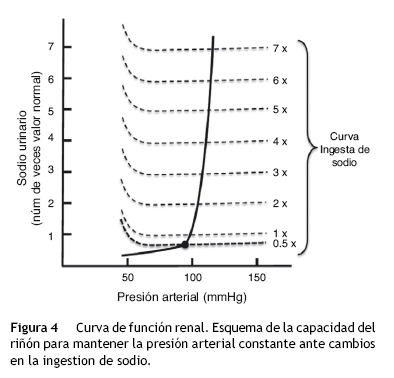
La figura 4 también establece que la TA está relacionada con la eliminación de NaU y con la ingesta de sal, de tal forma que si la ingesta es superior a la eliminación de Na y al gasto urinario, el volumen corporal se incrementa junto con la TA. Este hallazgo sugiere que en condiciones de balance, la presión arterial se encuentra exactamente en el mismo punto en que la eliminación de NaU y la ingesta de sal se encuentran en equilibrio16. Este punto de equilibrio se localiza gráficamente en el sitio donde ambas curvas se intersectan (fig. 4). Este punto puede ser utilizado para predecir el valor de TA a largo plazo. Cuando existe un valor de TA superior al punto de equilibrio, tanto la eliminación de NaU como el volumen urinario aumentan más que la ingesta de Na; por el contrario, cuando el valor de TA se encuentra por debajo del punto de equilibrio, se produce una mayor ingesta de Na en comparación con la eliminación de NaU y el volumen de orina.
La natriuresis de presión permite un control muy exacto de la TA y está regulado por el principio de retroalimentación de ganancia infinita16. Este mecanismo de control del volumen corporal siempre mantendrá la TA en su nivel basal (punto de equilibrio), independientemente de la magnitud y dirección del cambio temporal de las cifras de TA. Este principio de ganancia infinita se sustenta en 3 leyes17:
I) Es imposible modificar el control crónico de la TA a un valor distinto sin alterar primeramente el punto de equilibrio.
II) Si el punto de equilibrio se modifica, lo hará también de manera subsecuente, el nivel TA a largo plazo.
III) Las 2 principales determinantes del control de la TA a largo plazo son la curva de función renal y la curva de ingesta de Na, por lo tanto sólo aquellos factores que modifiquen alguna de estas variables tienen la capacidad de alterar la TA.
Curvas de función renal anormales
La curva de función renal que representa a la HSS es aquella en que la pendiente es menor con respecto a la curva normal y existe desviación hacia la derecha18 (fig. 5). Esta alteración traduce menor capacidad de excreción de NaU, defecto que se acompaña de cifras elevadas de la TA sistémica. Analizando esta gráfica resulta evidente que conforme se incremente la ingesta de Na, aumentará también la TA.
Algunos investigadores, entre ellos Kimura y Brenner19,20 consideran que este tipo de curva está determinada por el coeficiente de filtración (Kf) y la tasa de reabsorción tubular de Na, por lo que la disminución en el primero o aumento en la segunda, ocasionan HSS19,20; sin embargo, aunque el Kf y la reabsorción tubular son factores muy importantes, no son los únicos involucrados en la excreción de NaU ni en las anormalidades de la curva de natriuresis de presión. Se considera que alteraciones en la regulación del SRAA, la hiperactividad del sistema simpático (SNS) y la disminución de nefronas son los principales factores que modifican la curva de natriuresis de presión7.
Génesis de hipertensión sensible a sal
Recientemente, Johnson publicó una teoría que explica los mecanismos patogénicos que anteceden a la elevación de cifras de TA secundarios a ingesta abundante de sal14. Según su teoría, el desarrollo de HAS pasa a través de estadios en los cuales la TA se eleva episódicamente antes de mantenerse persistentemente alta.
Las cifras de TA oscilan durante el día, llegando a registrar presión sistólica de hasta 150mmHg21,22, incluso en sujetos catalogados como normotensos. Es probable que esta elevación transitoria de TA esté relacionada con hiperactividad del SNS inducida por factores genéticos, ambientales o familiares23, y activación del SRAA; los individuos que experimentan estos cambios de TA son los llamados sujetos hiperreactores24. Estas oscilaciones representan la primera fase de la HSS.
El mecanismo de autorregulación renal tiene como objetivo aumentar la resistencia de la arteriola aferente para evitar que las elevaciones de presión arterial sistémica sean transmitidas al glomérulo. La autorregulación comprende 2 factores principales: el mecanismo miogénico, propio de la mayoría de los lechos vasculares, y el mecanismo de retroalimentación túbulo glomerular25. La teoría de Johnson se fundamenta en que este mecanismo no es inmediato ni protege completamente14; al menos de manera transitoria, la HAS puede ser transmitida a los capilares peritubulares posglomerulares. Estos cambios hemodinámicos se asocian con incremento en la presión capilar peritubular26,27, y contrario a lo que se esperaría, no se acompanan de incremento de flujo sanguíneo local, por el contrario, hay evidencia de hipoperfusión renal, sobre todo a nivel medular y yuxtamedular28.
La exposición transitoria de los vasos sanguíneos renales a TA elevada en forma repetida y frecuente, facilita la transición hacia un estado de HAS persistente y desarrollo de sensibilidad a sal. Los capilares glomerulares, a diferencia de los posglomerulares están mejor adaptados a cambios de TA, debido a la existencia de podocitos y células mesangiales, estructuras que brindan soporte a la pared capilar14. Los capilares peritubulares al estar desprovistos de células musculares y/o pericitos, son más susceptibles de experimentar cambios estructurales secundarios a alteraciones hemodinámicas, como son la transformación pericapilar de fibroblastos a miofibroblastos29. Además de los cambios en los capilares peritubulares descritos previamente, existen otras alteraciones29: proliferación del músculo liso vascular (principalmente de la arteriola aferente)30, atrofia y dilatación tubular, fibrosis intersticial en parches, rarefacción capilar, presencia de osteopontina e infiltrado intersticial de linfocitos y macrófagos29. En conjunto, estas anormalidades se conocen como lesión túbulointersticial (TI).
Con ayuda de los postulados de Johnson se han explicado las diferencias de la respuesta encontrada, después de la disminución de Na en la dieta de sujetos jóvenes adultos y adultos mayores con hipertensión. En los primeros, el mecanismo de natriuresis de presión se encuentra íntegro y la HAS encontrada es resistente a sal; por el contrario, en los individuos mayores, en quienes la elevación repetida de la presión arterial ha producido el daño estructural de los capilares peritubulares, se observa incapacidad para eliminar eficientemente el Na a través de la orina. La HAS de estos sujetos es HSS30.
Lesión intersticial renal e hipertensión sensible a sal
En estudios previos, Lombardi31 infundió dosis presoras de AngII a ratas durante 2 semanas, produciendo HAS y proteinuria. Después de la suspensión del péptido, como era de esperarse, la TA elevada regresaba a valores cercanos a lo normal y la función renal se recuperaba. El análisis estructural corroboró los cambios histológicos comentados en párrafos anteriores. La segunda fase de estos experimentos consistió en someter a las ratas a dieta baja y alta en sal (NaCl al 0.1% y 4%, respectivamente) durante 6 semanas. A las 3 semanas de iniciada la dieta, los animales con ingesta alta en sal desarrollaron nuevamente HAS, no así los animales alimentados con una dieta baja en Na. De esta forma se corroboró que las alteraciones TI son capaces de inducir HSS31.
La lesión estructural que induce HSS en los modelos animales se produce no sólo con AngII sino también con la administración de fenilefrina32, ciclosporina33, desoxicorticosterona37 e inhibidores de la síntesis de óxido nítrico (NO)30,34. Algunos estudios reportan que el grado de HAS se correlaciona con el grado de lesión TI (r2=0.744)32.
El infiltrado TI renal es un hallazgo universal en los modelos animales de HSS y un elemento de importancia crítica en la desviación hacia la derecha de la curva de natriuresis de presión35, por lo que algunos investigadores como Rodríguez-Iturbe, sugieren que la disminución del infiltrado TI con compuestos que eviten la acumulación de células inflamatorias en el riñón previene el desarrollo de HSS36.
La importancia del infiltrado TI en la patogenia de la HSS se puso en evidencia por primera vez por Svendsen37 hace casi 3 décadas, al observar que la HSS después de la administración de desoxicorticosterona no se presentaba en ratones sin timo y que los injertos de este órgano restauraban la hipertensión dependiente de sal. Estos resultados fueron corroborados de manera contundente por Guzik38 al infundir AngII durante 3 semanas a ratas, cuyas concentraciones de linfocitos B y T fueron modificadas mediante ingeniería genética. Cuando la AngII era administrada a animales carentes de linfocitos B y T, no se presentaba hipertensión arterial. Si la maniobra se repetía en ratas con mayor población de linfocitos B, la respuesta era la misma, es decir normotensión. Por el contario, cuando el experimento se realizaba en ratas con presencia de linfocitos T se observaba desarrollo de HAS38.
Atenuación del infiltrado intersticial renal y disminución de la tensión arterial
Con la finalidad de establecer el papel del infiltrado intersticial en la génesis de HSS, Quiroz39 y Rodríguez-Iturbe40 observaron la respuesta tanto de la TA como del infiltrado TI encontradas con la administración de mofetil micofenolato (MMF, inmunosupresor que regula la síntesis de novo de purinas e inhibe la proliferación linfocitaria, así como la producción de moléculas de adhesión41) en un modelo de HSS con NG-Nitro-L-Arginina Metil Ester (L-NAME, inhibidor de la síntesis de óxido nítrico)39 y con AngII40.
La exposición de la rata a la inhibición de la producción de NO mediante L-NAME incrementó la TA, incluso en presencia de MMF. Sin embargo, una segunda fase de alimentación alta en sal tras la suspensión de este fármaco, no producía la HAS característica39. Este estudio concluyó que MMF evitó el desarrollo de HSS. Observaciones posteriores hallaron que la administración de este inmunosupresor no disminuyó la lesión TI característica de la infusión de L-NAME (definida como dilatación tubular, formación de cilindros y/o expresión de osteopontina por parte de los túbulos), pero sí disminuyó significativamente el infiltrado de macrófagos, linfocitos T, linfocitos T activados y linfocitos T que expresan AngII39.
La importancia del infiltrado TI de linfocitos T y macrófagos, así como su relación con la TA en un modelo de HSS se puso en evidencia por Franco42 mediante experimentos con técnicas de microdiálisis: después de alimentar a ratas con dieta alta en sal, los niveles plasmáticos de AngII tenían una correlación negativa (p<0.0001) con la TA. Por el contrario, la concentración intrarrenal de AngII, el infiltrado TI de linfocitos T - macrófagos y la TA tuvieron una correlación positiva (p<0.0001)42.
El efecto de MMF sobre la TA, por razones éticas ha sido difícil de evaluar en grandes cohortes de pacientes hipertensos; sin embargo, estudios clínicos con un número pequeño de pacientes sugieren que la disminución del infiltrado TI tiene traducción clínica: en un grupo de 8 pacientes hipertensos con diagnóstico de artritis reumatoide o psoriasis, con indicación para iniciar MMF como parte de su tratamiento, Herrera43 documentó menor TA durante los 3 meses de duración del tratamiento inmunosupresor, con elevación ulterior de la misma luego de su suspensión.
El mecanismo mediante el que MMF normaliza la TA en modelos de HSS se conoce de manera parcial. Franco y Rodríguez-Iturbe evaluaron la curva de función renal de ratas con HSS tratadas con MMF (información no publicada). Los animales tratados con MMF presentaron una concentración urinaria y fracción excretada Na muy similares a los del grupos control (ratas no hipertensas), demostrando que la disminución del infiltrado TI normaliza la curva de función renal; dicho en otras palabras MMF normaliza la natriuresis de presión.
Conclusiones
Los mecanismos moleculares que asocian la respuesta de la TA a la inflamación no se han determinado, sin embargo la información derivada de estos estudios abre nuevas alternativas para el desarrollo de fármacos que controlen eficazmente el desarrollo y las complicaciones asociadas a la HAS, enfermedad considerada actualmente como un problema de salud pública. Conocer los mecanismos patogénicos de la HAS es de suma utilidad para todo el personal médico involucrado en el manejo de estos pacientes y permite planear estrategias terapéuticas encaminadas a mejorar el estado de salud de estos enfermos.
Financiamiento
No se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Bibliografía
1. Blumenfeld JD, Laragh JH. Primary and Secondary Hypertension. En: Brenner BM, editor. Brenner and Rector's The Kidney. Philadelphia: Elsevier; 2008. p. 1465-528. [ Links ]
2. Barquera S, Campos-Nonato I, Hernández-Barrera L, et al. Hypertension in Mexican adults: Results from the National Health and Nutrition Survey 2006. Salud Pública Mex. 2010;52 Suppl. 1:S63-71. [ Links ]
3. Van Vliet BN, Montani JP. The time course of salt-induced hypertension, and Why It Matters. Int J Obes. 2008;32:S35-47. [ Links ]
4. Weinberg MH, Fineberg NH. Sodium and volume sensitivity of blood pressure. Age and pressure change over time. Hypertension. 1991;18:67-71. [ Links ]
5. Midgley JP, Matthew AG, Greenwood CMT, et al. Effect of reduced dietary sodium on blood pressure: a meta-analysis of randomized controlled trials. JAMA. 1996;275:1590-7. [ Links ]
6. Rodríguez-Iturbe B, Romero F, Johnson RJ. Pathophysiological mechanism of salt sensitive hypertension. Am J Kidney Dis. 2007;50:655-72. [ Links ]
7. Franco M, Sanchez-Lozada LG, Bautista R, et al. pathophysiology of salt-sensitive hypertension: a new scope of an old problem. Blood Purif. 2008;26:45-8. [ Links ]
8. Eaton DC, Pooler JP. Control of sodium and water excretion. Regulation of plasma volume and plasma osmolality and renal control of systemic blood pressure. En: Eaton DC, Pooler JP, editores. Vander's Renal Physiology. Atlanta: McGraw-Hill; 2006.p. 98-133. [ Links ]
9. Guyton A, Hall J, Coleman T, et al. The dominant role of the kidneys in long-term arterial pressure regulation in normal and hypertensive states. En: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis, and Management, 2. New York: Raven Press; 1995. p. 1311-26. [ Links ]
10. Guyton AC, Hall JE. Dominant role of the kidneys in long-term regulation of arterial pressure an in hypertension: the integrated system for pressure control. En: Guyton AC, Hall JE, editores. Textbook of Medical Physiology. 10th ed. Philadelphia: WB Saunders; 2000. p. 221 -34. [ Links ]
11. Hall JE, Jackson TE. The basic kidney - blood volume -pressure regulatory system. The pressure diuresis and natriuresis phenomena. En: Guyton AC, editor. Arterial Pressure and Hypertension, Circulatory Physiology. 1st ed. Philadelphia: WB Saunders; 1980. p. 87 -99. [ Links ]
12. Navar LG. The kidney in blood pressure regulation and development of hypertension. Med Clin North Am. 1997;81:1165-98. [ Links ]
13. Nakamura T, Alberola AM, Salazar FJ, et al. Effects of renal perfusion pressure on renal interstitial hydrostatic pressure and Na+ excretion: role of endothelium derived nitric oxide. Nephron. 1998;78:104-11. [ Links ]
14. Johnson RJ, Schreiner JF. Hypothesis: The role of acquired tubulointerstitial disease in the pathogenesis of salt-sensitive hypertension. Kidney Int. 1997;52:1169-79. [ Links ]
15. Navar LG. Regulation of renal hemodynamics. AdvPhysiol Educ. 1998;20:S221 -35. [ Links ]
16. Norman RA, Cowley AW, DeClue JW. The renal function curve and its significance in long-term arterial pressure regulation. En: Guyton AC, editor. Arterial Pressure And Hypertension, Circulatory Physiology. 1st ed. Philadelphia: WB Saunders; 1980. p. 100 -16. [ Links ]
17. Coleman TG. Infinite gain feature of the kidney - blood volume -pressure regulator; the three laws of long-term arterial pressure regulation; renal servocontrol of arterial pressure. En: Guyton AC, editor. Arterial pressure and hypertension, Circulatory Physiology. 1st ed. Philadelphia: WB Saunders; 1980. p. 117 -25. [ Links ]
18. Kimura G, Brenner BM. The renal basis for salt sensitivity in hypertension. En: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis, and Management, Vol. 2. New York: Raven Press; 1995. p. 1569 -86. [ Links ]
19. Kimura G, Brenner BM. A method for distinguishing salt-sensitive from non-salt-sensitive froms of human and experimental hypertension. Curr Opin Nephrol Hypertens. 1993;2:341-9. [ Links ]
20. Gong R, Dworkin LD, Brenner BM. The renal circulation and glomerular ultrafiltration. En: Brenner BM, editor. Brenner and Rector's The Kidney. 8th ed. Philadelphia: Elsevier; 2008. p. 91 -120. [ Links ]
21. Littler WA, Honour AJ, Sleight P. Direct arterial pressure and electrocardiogram during motor car driving. Br Med J. 1973;2:273-7. [ Links ]
22. Williamson OK. Arterial blood pressure records before and after muscular exertion. Br Med J. 1909;1:530. [ Links ]
23. Julius S. The evidence for a pathophysiologic significance of the sympathetic overactivity in hypertension. Clin Exp Hypertens. 1996;18:305-21. [ Links ]
24. Watson RD, Stallard TJ, Flinn RM, et al. Factors determining direct arterial pressure and its variability in hypertensive man. Hypertension. 1980;2:333-41. [ Links ]
25. Franco M. Mecanismos de autorregulación renal. Principia Cardiológica. 1994;3:59 -70. [ Links ]
26. Myers BD, Deen WM, Brenner BM. Effects of norepinephrine and angiotensin ii on the determinants of glomerular ultrafiltration and proximal tubule fluid reabsorption in the rat. Circ Res. 1975;37:101-10. [ Links ]
27. Cowley AW, Mattson DL, Lu S, et al. The renal medulla and hypertension. Hypertension. 1995;25:663-73. [ Links ]
28. Faubert PF, Chou SY, Porush JG. Regulation of papillary plasma flow by angiotensin II. Kidney Int. 1987;32:472-8. [ Links ]
29. Johnson RJ, Alpers RE, Yoshimura A, et al. Renal injury from angiotensin II-mediated hypertension. Hypertension. 1992;19:464-74. [ Links ]
30. Johnson RJ, Herrera-Acosta J, Schreiner GF, et al. Subtle acquired renal injury as a mechanism of salt sensitive hypertension. N Engl J Med. 2002;346:913-23. [ Links ]
31. Lombardi D, Gordon KL, Polinsky P, et al. Salt-sensitive hypertension develops after short-term exposure to angiotensin II. Hypertension. 1999;33:1013-9. [ Links ]
32. Johnson RJ, Gordon KL, Suga S, et al. Renal injury and salt-sensitive hypertension after exposure to catecholamines. Hypertension. 1999;34:151-9. [ Links ]
33. Kang DH, Kim YG, Andoh TF, et al. Post-cyclosporin-mediated hypertension and nephropathy: amelioration by vascular endothelial growth factor. Am J Physiol Renal Physiol. 2001;280:F727 -36. [ Links ]
34. Ribeiro MO, Antunes E, de Nucci G, et al. Chronic inhibition of nitric oxide synthesis. a new model of arterial hypertension. Hypertension. 1992;20:298-303. [ Links ]
35. Rodríguez-Iturbe B. El papel de la infiltración renal de células inmunocompetentes en la patogenia de hipertensión arterial. Nefrología. 2008;28:483-92. [ Links ]
36. Rodríguez-Iturbe B. Renal infiltration of immunocompetent cells: cause and effect of sodium-sensitive hypertension. Clin Exp Nephrol. 2010;14:105-11. [ Links ]
37. Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus-dependent phase of doca and salt hypertension in mice. Acta Path Microbiol Scand. 1976;84:523-8. [ Links ]
38. Guzik TJ, Hoch NE, Brown KA, et al. Role of t cells in the genesis of angiotensin ii-induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449-60. [ Links ]
39. Quiroz Y, Pons H, Gordon KL, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol. 2001;281:F38-47. [ Links ]
40. Rodríguez-Iturbe B, Pons H, Quiroz Y, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int. 2001;59: 2222 -32. [ Links ]
41. Allison AC, Eugui RM. Mycophenolate mofetil and its mechanism of action. Immunopharmacology. 2000;47:85-118. [ Links ]
42. Franco M, Martínez F, Quiroz Y, et al. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;293: R251 -6. [ Links ]
43. Herrera J, Ferrebuz A, García-McGregor E, et al. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17:S218-25. [ Links ]

