










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos de cardiología de México
On-line version ISSN 1665-1731Print version ISSN 1405-9940
Arch. Cardiol. Méx. vol.79 suppl.2 Ciudad de México Dec. 2009
PARTE II
Investigación Clínica
Alteraciones plaquetarias en la diabetes mellitus tipo 2
Platelet abnormalities in type 2 diabetes mellitus
Cuauhtémoc Matadamas–Zárate,1 Julia Hernández–Jerónimo,2 Eduardo Pérez–Campos,3 Abraham Majluf–Cruz4
1 HGZ No. 1. IMSS. Facultad de Medicina. UABJO. Oaxaca, Oax.
2 Facultad de Medicina. UABJO. Oaxaca, Oax.
3 CICIMEBIO. Facultad de Medicina. UABJO. Oaxaca, Oax.
4 Unidad de Investigación Médica en Trombosis, Hemostasia y Aterogénesis. HGR Gabriel Mancera. IMSS. México, D. F.
Correspondencia:
Dr. Abraham Majluf Cruz.
Apartado Postal 12–1100. México 12, D. F., México.
Teléfono y fax: 0155 5639 5822, extensión 20855.
Correo electrónico: amajlufc@gmail.com
Recibido el 18 de octubre de 2007;
Aceptado el 20 de mayo de 2009.
Resumen
La diabetes mellitus es un grave problema de salud mundial. Las complicaciones vasculares son la principal causa de morbimortalidad. El diabético tiene una evolución aterotrombótica acelerada y peor que la de otras entidades clínicas; pese a ello, la hiperglucemia per se no explica por completo las complicaciones isquémicas que se observan en estos enfermos. La mayoría de los eventos isquémicos arteriales se precipitan por rotura de la placa ateroesclerótica, activación plaquetaria y la trombosis resultante. En la diabetes se producen diversas alteraciones del sistema de coagulación, como disfunción endotelial, hiperactividad plaquetaria, generación de trombina y fibrinólisis disminuida, eventos patológicos que favorecen la trombosis. La plaqueta es clave en la aterotrombosis diabética debido a que existe hipersensibilidad plaquetaria a agonistas y respuesta baja a los agentes antiplaquetarios terapéuticos, además de hiperactividad plaquetaria en sitios de daño endotelial, hiperagregabilidad, resistencia a los efectos inhibitorios de la insulina y una producción endotelial baja de prostaciclina y óxido nítrico. Estas alteraciones dependen del medio ambiente "tóxico" (hiperglucemia) o son intrínsecas a la plaqueta. Por todo lo anterior, la plaqueta es otro blanco de los efectos deletéreos de la resistencia a la insulina. Ya que la plaqueta es clave en la ateroesclerosis y en las complicaciones vasculares de la diabetes mellitus, esta revisión analiza las alteraciones plaquetarias características de esta enfermedad metabólica.
Palabras clave Plaquetas; Diabetes mellitus tipo 2; Aterotrombosis; México.
Abstract
Diabetes mellitus is a problem of health worldwide being vascular complications the main causes of morbidity and mortality in this population. Diabetics have a fast atherothrombotic evolution which is worse than that observed for other clinical entities; however, hyperglycemia itself may not totally explain the ischemic complications observed in these patients. Most ischemic arterial events are precipitated by plaque rupture, platelet activation, and thrombosis. Several abnormalities in the blood coagulation system have been described associated to diabetes mellitus, all of them predisposing to thrombosis: endothelial cell dysfunction, platelet hyperreactivity, thrombin generation and hypofibrinolysis. Platelets play a key role in diabetic atherothrombosis due to platelet hypersensitivity to physiological agonists, low response to therapeutical antiplatelet agents, platelet hyperreactivity in sites of endothelial cell damage, hyperaggregability, resistance to the inhibitory effects of the insulin, and low endothelial production of prostacyclin and nitric oxide. All these phenomena have been associated to either a toxic microenvironment due to hyperglycemia or to intrinsic platelet abnormalities. Based on all these facts, it is proposed that platelets may be another target for the negative effects of insulin–resistance state. Because platelets are crucial in the atherosclerotic process and in the genesis of the vascular complications of diabetes mellitus, this review analyses the platelet abnormalities observed in this metabolic disease.
Key words: Platelets; Type 2 diabetes mellitus; atherothrombosis; Mexico.
Introducción
La diabetes mellitus (DM) es uno de los problemas de salud más importantes en el mundo. En México su prevalencia aumentó de 7.2% entre los 20 y 65 años en 1993 hasta 11.8% en la última década. Hoy se detectan 180 mil casos nuevos/año. En 2002, fue la primera causa de muerte: 54 828 defunciones (12% del total).1 Casi 90% de los diabéticos que debutan después de los 30 años tiene diabetes mellitus tipo 2 (DM–2).2 Las complicaciones micro y macrovasculares son la principal causa de morbimortalidad. La enfermedad macrovascular (enfermedad arterial coronaria [EAC], enfermedad vascular cerebral [EVC] y enfermedad arterial periférica [EAP]), aumenta dos a cuatro veces en la DM–2 comparada con los sujetos no diabéticos.3 La EAC y su mayor expresión, la cardiopatía isquémica (angina inestable y síndromes coronarios agudos), es la primera causa de muerte prematura en la DM–2. Casi 75% de los diabéticos fallece por cardiopatía o infarto cerebral. Además del riesgo elevado de EAC, el diabético tiene un peor pronóstico en la evolución de la aterotrombosis;4 el que sufre un infarto agudo de miocardio (IAM) tiene más complicaciones hospitalarias y mayor recurrencia de isquemia comparado con el no diabético: 45% vs. 19%, respectivamente.5
La causa de las complicaciones macrovasculares de la DM–2 no es clara. La hiperglucemia crónica no explica del todo estas complicaciones, a diferencia del daño microvascular. En la DM–2, la ateroesclerosis y la trombosis se sinergizan para elevar el riesgo cardiovascular. La mayoría de los eventos isquémicos coronarios y cerebrales ocurre por oclusión vascular secundaria a la rotura de la placa ateroesclerótica, la activación plaquetaria y la trombosis resultante. varios sistemas que mantienen la integridad y función vascular se alteran en la DM. Las alteraciones plaquetarias funcionales y la hipercoagulabilidad resultante afectan la DM y el síndrome metabólico (SM); es posible que en este ambiente dismetabólico la hiperfunción plaquetaria tenga diversas vías patógenas.
Fisiología plaquetaria
Las plaquetas forman el tapón hemostático (hemostasia primaria), aportan una superficie hemostática y factores hemostáticos y proangiógenos.6 Al activarse, cambian de forma, se adhieren al subendotelio, secretan el contenido de sus gránulos y se agregan en el coágulo. sus activadores son trombina, colágena y adrenalina (exógenos a la plaqueta) y ADP y tromboxano A2 (TxA2) sintetizados en la plaqueta.7 La adhesión al vaso dañado ocurre en una fase de contacto y una de diseminación sobre el subendotelio. La lesión endotelial expone colágena subendotelial I y III que permiten la adhesión, la cual, bajo un flujo sanguíneo rápido (en la arteria), requiere factor de von Willebrand (FvW) y la glucoproteína plaquetaria Ib (GPIb). La interacción FvW–GPIb activa la plaqueta para que exprese el receptor GPIIb/IIIa, sitio de unión para proteínas adherentes como el fibrinógeno. El GPIIb/IIIa permite que la plaqueta interactúe con el FvW, que se disemine sobre el subendotelio y se agregue. En la agregación, la plaqueta secreta factores hemostáticos y de crecimiento para reparar la lesión y el fibrinógeno se une a la GPIIb/IIIa para formar puentes interplaquetarios.
Activación plaquetaria
Inicia en su superficie al interactuar un agonista con su receptor, desencadenar la transducción de la señal y culminar en diversas funciones. En la membrana, el GPIIb/ IIIa cambia de forma y expone el sitio de unión al fibrinógeno.8 Para que el complejo agonista–receptor inicie la formación de segundos mensajeros, se requieren proteínas g que regulan la concentración de cAMP (mediante el estímulo o la inhibición de la adenilatociclasa), la función de los canales del calcio (Ca) y el potasio, la hidrólisis de fosfatidilcolina y la activación de las fosfolipasas A2 (PLA2) y C (PLC).6
Transducción de la señal, sistema de segundos mensajeros
Los agonistas estimulan vías que generan moléculas activas o segundos mensajeros. Dos de éstas se inician con la hidrólisis de fosfolípidos de la membrana y son clave en la activación: la vía de los inositoles o de la PLC y la vía del ácido araquidónico (AA) o de la PLA2. La acción de los agonistas sobre la adenilatociclasa aumenta o disminuye el cAMP, y con ello inhiben o estimulan la activación.9
Vía de los inositoles. La hidrólisis de los inositoles se inicia cuando la PLC metaboliza al fosfatidil–inositol (PI) y otros fosfolípidos sin aumentar el Ca citosólico.10 se forman segundos mensajeros: trifosfato de inositol (IP–3), DAg y ácido fosfatídico. El IP–3 libera Ca del sistema tubular denso (SfTD) y eleva su concentración citosólica.10 El Ca activa enzimas intraplaquetarias. El DAG es un cofactor que activa a la PKC, que fosforila proteínas.
Vía del AA. El metabolismo del AA requiere PLA2.11 En la plaqueta, la ciclooxigenasa (COX) oxida el AA hasta prostaglandinas (PG) y tromboxanos (Tx).12 En la plaqueta, la sintetasa de tromboxano (TS) genera TxA2; en el endotelio, la sintetasa de prostaciclina forma prostaciclina (PGI2). El TxA2, un proagregante potente, estimula la contracción muscular lisa vascular y respiratoria.13 La COX y la TS se localizan en el STD, donde se sintetiza TxA2. El ácido acetilsalicílico inactiva la COX en forma irreversible.
Calcio y proteincinasas. El Ca es un segundo mensajero que afecta la actividad plaquetaria global.14 La plaqueta en reposo mantiene el Ca citosólico libre bajo por medio de bombas que lo sacan al exterior o lo introducen en el STD.15 En la plaqueta activa, el Ca aumenta ya que el IP–3 libera Ca del STD y porque otra parte entra a través de la membrana plasmática.16 El aumento del Ca induce el cambio en la GPIIb/IIIa. Al aumentar el Ca, también se activan la PLA2 y la PLC.
Agonistas plaquetarios y sus receptores.17 La colágena y la trombina son los agonistas más potentes in vivo. El ADP, la serotonina, el factor activador de plaquetas (PAF) y el TxA2 son amplificadores.18 La colágena y la trombina estimulan la hidrólisis de inositoles, la formación de eicosanoides, la secreción y agregación que se bloquean de modo parcial con inhibidores del TxA2 y elevan el Ca libre.19 Esto indica que sus receptores se acoplan a la PLA2 y a la PLC. Los agonistas débiles se ligan más a la PLA2 y a la formación de TxA2.20
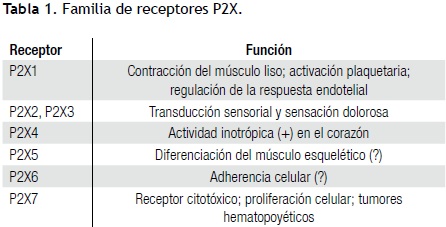
Receptores plaquetarios para el ADP. El ADP se une a la plaqueta por tres receptores purinérgicos que inducen la fosforilación proteínica, formación de eicosanoides, aumento de Ca libre y expresión del receptor de fibrinógeno.21 Así, se relaciona con la agregación y secreción. En la activación por ADP intervienen la inhibición de la adenilciclasa, el aumento del Ca y su movilización vía PLC. Hay dos subfamilias de receptores para ADP: P2X y P2Y22,23 (Tabla 1). El P2X1 es crucial en la agregación plaquetaria en medio de flujo sanguíneo rápido. Existen transcritos de P2X1 en megacariocitos y plaquetas,24 pero el P2X1 funcional se expresa en plaquetas, donde quizá es el factor determinante del ingreso rápido de Ca en la plaqueta expuesta a ADP y de la agregación inducida por colágena.25 La subfamilia de los receptores P2Y (P2YR) comprende ocho tipos.21 El ADP es crucial en la función plaquetaria al activar P2Y1 y P2Y12. El P2Y1 inicia el cambio de forma y la agregación al movilizar Ca del sTD. El P2Y12 permite la agregación completa y su estabilización. El P2Y1 es el receptor de ADP al inicio de la adhesión y activación plaquetarias;26 es clave para fijar la plaqueta al FvW e influenciar la liberación inicial de Ca del STD y el flujo transmembrana luego que la adhesión concluyó.6 Los P2YR activos inhiben la generación de cAMP e inducen una conformación idónea de la GPIIb/IIIa para la agregación.27 La clonación y expresión de los receptores P2X y P2Y son la base de nuevas terapias.28 Ticlopidina y clopidogrel son antagonistas irreversibles de P2Y1
Receptores plaquetarios para adrenalina. son receptores adrenérgicos alfa2. La adrenalina es el único agonista que induce agregación sin cambio de forma. Al igual que la trombina, inhibe la adenilciclasa y la formación de cAMP y activa la PLA2. Produce un efecto sinérgico ya que potencia el efecto de concentraciones subumbrales de otros agonistas.29
El sistema de coagulación y su contribución a la aterogénesis de la DM–2
La DM–2 cursa con ateroesclerosis acelerada y su patogenia incluye varios mecanismos relacionados con el sistema de coagulación: disfunción endotelial (Tabla 2), hiperactividad plaquetaria, generación intravascular de trombina y fibrinólisis disminuida.30,31 El resultado es un desequilibrio hemostático que favorece la trombosis, por lo que la DM–2 es un estado protrombótico o trombofílico.32 La trombosis es crucial en la aterogénesis, la plaqueta es clave en la trombosis arterial y, por lo tanto, en la aterotrombosis de la DM. In vitro se conocen alteraciones como hipersensibilidad plaquetaria a los agonistas y respuesta baja a los antiplaquetarios terapéuticos, efectos que inciden en la génesis de la ateroesclerosis al aumentar la actividad plaquetaria en sitios de daño endotelial. La disfunción plaquetaria característica de la DM–2 (hiperagregabilidad y resistencia a la inhibición de la insulina) aunada a la caída de la producción endotelial de PGI2 y NO, potencian la aterotrombosis y el riesgo de trombosis.8 A pesar de la evidencia del desempeño plaquetario en la vasculopatía de la DM–2, se desconoce la bioquímica de su hiperactividad. Un enigma es si la activación plaquetaria persistente de la DM–2 se debe a la mayor prevalencia de lesiones ateroescleróticas o si es consecuencia de las alteraciones metabólicas y hemodinámicas propias de la DM sobre la bioquímica plaquetaria. Ya que la disfunción plaquetaria se demuestra tanto en el plasma rico en plaquetas como en suspensión de plaquetas lavadas,30 es factible la combinación de mecanismos generados por el medio ambiente "tóxico" (la hiperglucemia) con otros intrínsecos de la plaqueta en las vías de señalización o en los receptores del ADP o de la adrenalina.33 La disfunción plaquetaria de la DM puede relacionarse con la disfunción endotelial, el estrés oxidativo y las alteraciones metabólicas, las cuales parecen tener un papel protagónico.31

Disfunción plaquetaria
La plaqueta es una protagonista importante en la aterogénesis. La concentración de glucosa intraplaquetaria es cercana a la extracelular ya que la entrada de glucosa a la plaqueta no depende de insulina.8 La hiperglucemia crónica produce cambios en la bioquímica y fisiología plaquetarias y contribuye a la hiperactividad plaquetaria propia de la DM–2.34 Hay hipersensibilidad plaquetaria a los agonistas e hiposensibilidad a los antiplaquetarios, dos efectos proaterógenos que elevan la actividad plaquetaria en sitios de daño endotelial (Tabla 3). La disfunción plaquetaria (hiperactividad y resistencia a la inhibición por la insulina), más una producción endotelial menor de PGI2 y NO magnifican la respuesta aterógena y elevan el riesgo aterotrombótico de la DM–2.8 En el diabético aumentan algunos marcadores de activación plaquetaria que evalúan el daño vascular: CD–62P, CD–63, PAC–1, anexina 5 y PDMP.35 Así, en potencia, en la DM–2 pueden ocurrir alteraciones en casi todos los mecanismos que regulan la función plaquetaria, aunque existen diferencias entre los dos principales tipos de diabetes36–38 (Tabla 4).


Respuesta a agonistas La respuesta a adrenalina, ADP, colágena y trombina aumenta en las plaquetas en la DM, pero esta hiperfunción es difícil de demostrar en pacientes controlados.8 Es factible que la alteración exista en una vía común de la activación plaquetaria, la vía del AA; sin embargo, in vitro, la hiperactividad plaquetaria inducida por trombina persiste después que la vía del AA se inhibe,31 lo que sugiere la existencia de otros mecanismos de activación plaquetaria en la DM. La hiperactividad parece independiente de la vía del ADP39 y no disminuye después de siete días de insulinización, la cual normaliza la glucosa sanguínea pero no el perfil de lípidos. se sugiere que en la DM hay un círculo vicioso: el daño vascular conduce al plaquetario y la disfunción plaquetaria contribuye a la enfermedad vascular, aunque es factible que el daño vascular no cause hiperactividad plaquetaria. En animales con DM, el aumento en la actividad plaquetaria y de la síntesis de TxA2 se detectó a los pocos días que se hicieron diabéticos con estreptozotocina, antes que la vasculopatía fuera evidente,40 lo que hace factible una alteración plaquetaria intrínseca en la DM. La DM aumenta la síntesis plaquetaria de TxA2 y el control metabólico la disminuye.41 La alteración metabólica más que la enfermedad vascular es la que genera la actividad plaquetaria persistente.
Hidrólisis del fosfatidil–inositol. Aunque el estudio de la alteración plaquetaria en la DM se centra en la vía del AA, hay otros mecanismos que explican la hipersensibilidad plaquetaria a sus agonistas, por ejemplo la vía del PI. La hidrólisis del PI, un hecho inicial en la activación plaquetaria, aumenta en las plaquetas hiperactivas en la DM–2, pero disminuye en la DM–1. Quizá, la hipersensibilidad a los agonistas propia de la DM–2 se acompañe de recambio alto de PI. Además, por la naturaleza multifactorial de la hiperactividad plaquetaria en la DM, la menor actividad del ciclo del PI puede compensar la hiperactividad de otras vías.8 La hiperactividad plaquetaria del envejecimiento se asocia con mayor hidrólisis del PI, hecho compatible con que la hiperactividad se relaciona con alteraciones de la vía del PI.42
Homeostasis del Cay el magnesio. El Ca plaquetario es necesario para el cambio de forma, secreción, agregación y producción de TxA2. su homeostasis se altera en la DM ya que aumenta su liberación, lo que genera hiperactividad plaquetaria. En la DM–2, la hiperactividad plaquetaria se asocia con aumento de Ca.43 su movilización aumenta desde el retículo endoplásmico. En la DM–1 mal controlada (HbA1c > 8%), las plaquetas en reposo presentan más Ca intracelular que las de los controles. La concentración de Ca inducida por trombina aumenta sólo en pacientes controlados y sin complicaciones.44 Por el contrario, el diabético muestra concentraciones de Ca mayores que los controles.45 El cambio de dirección de la bomba Na+/Ca2+ aumenta el Ca intraplaquetario en la DM; la hiperglucemia prolongada induce cambios similares, lo que sugiere que participa en la hiperactividad plaquetaria de la DM.31 La hipermovilización de Ca desde el STD va seguida de hiperactividad plaquetaria. La caída del magnesio intracelular libre predice trombosis plaquetaria en correlación con la glucosa sanguínea, colesterol total, apolipoproteína B, hipertensión arterial, resistencia a la insulina (RI) e hipertrofia cardiaca.46
Productos finales de la glucosilación avanzada. son productos terminales de la reacción no enzimática entre la función aldehídica de la glucosa y los grupos amino libres proteínicos. En la DM, se acumulan en los tejidos a mayor velocidad. En presencia de hierro o cobre, las proteínas glucosiladas dan un electrón al oxígeno molecular para generar radicales de oxígeno. si la vida media de la proteína es mayor de 10 semanas, su fracción glucosilada se modifica de manera irreversible y resulta en PFGA o cuerpos de Maillard.47 La hiperglucemia genera glucosilación no enzimática de las proteínas de la membrana plaquetaria e induce cambios en su estructura, lo que modifica su permeabilidad selectiva y genera una dinámica anormal de los lípidos.31 Permite también la sobreexpresión de receptores plaquetarios (P–selectina y GPIIb/IIIa) y su interacción mayor con sus ligandos (fibrinógeno). La hiperglucosilación proteínica subendotelial con PFGA puede inactivar la producción de NO y disminuir con ello la inhibición plaquetaria.48
Plaquetas, dislipidemia y DM–2. El perfil lipídico característico de la DM–2 es un aumento de LDL (en primer lugar de LDL glucosiladas [GlycLDL] y de partículas LDL pequeñas y densas) y triglicéridos (TG) y disminución del HDL–C. Este perfil altera la función plaquetaria al interferir con la permeabilidad de la membrana y de los sistemas intracelulares. Las LDL pequeñas y densas son más susceptibles a oxidarse que las normales y las oxidadas inhiben la expresión de la NOs plaquetaria. La hiperglucemia aumenta la glucosilación no enzimática de LDL, que son más susceptibles al estrés oxidativo y a formar PFGA, lo que facilita la peroxidación lipídica. En la plaqueta, las GlycLDL elevan la concentración de Ca. El diabético tiene concentraciones plasmáticas altas de compuestos capaces de reaccionar con el ácido tiobarbitúrico, lo que sugiere peroxidación lipídica in vivo.31
Aumento en la expresión de GP. El diabético tiene una población alta de plaquetas activadas que expresan un mayor número de adhesinas como GPIb, GPIIb/IIIa, GP–53 lisosómico y P–selectina. En la DM–2, la hiperexpresión de GPIIb/llla se acompaña de una mayor unión al fibrinógeno e hiperagregabilidad. Además de reflejar hiperagregabilidad, la hiperexpresión de estas adhesinas sugiere que plaquetas y leucocitos se comunican para intervenir en el daño vascular inflamatorio. Para terminar, el fibrinógeno aumenta en muchos pacientes con DM–1 y DM–2.8
Plaquetas, síndrome metabólico y resistencia a la insulina
La asociación de obesidad o sobrepeso con perfil metabólico alterado se documentó hace 50 años. El síndrome metabólico (SM) es un grupo de factores de riesgo independientes que aparecen de manera simultánea en un individuo y lo predisponen a ateroesclerosis, trombosis y DM.49 El estado protrombótico es parte de su fisiopatología.8 Los componentes primarios (obesidad, dislipidemia, hipertensión arterial) están relacionados, dependen de múltiples sistemas fisiológicos y exhiben una etiología multifactorial compleja. Los obesos tienen 50 a 100% de aumento del riesgo de muerte prematura comparados con sujetos con IMC de entre 20 y –25 kg/m2. La obesidad (sobre todo la intraabdominal) aumenta la resistencia a la insulina (RI) al elevar la insulina.50 Los adipocitos viscerales afectan la función de la célula beta, la producción de glucosa hepática, la captación muscular de glucosa, la regulación del apetito y la inflamación mediada por adipocinas (lipasa de lipoproteína, leptina, resistina, IL–6, TNF–α y adiponectina).51 La grasa intraabdominal resiste más la supresión de la lipólisis de la insulina que la grasa subcutánea por aumento de TNF–α e IL–6.52 La dislipidemia aterógena (aumento de Tg y partículas pequeñas y densas de LDL y HDL–C bajo) se asocia a RI y DM, independientemente del colesterol de LDL; la mayoría de pacientes con RI presenta este fenotipo aun sin padecer DM y puede preceder a la DM por años. La dislipidemia aterógena y la RI guardan una relación metabólica. El otro factor diagnóstico en el SM es la hipertensión arterial, factor de riesgo para enfermedad macrovascular que ocurre en casi 30% de los casos con SM. La RI genera hipertensión arterial y afecta de manera directa la señalización vascular y la función endotelial.53
La definición de RI que sólo considera la insuficiencia de esta hormona no explica todos sus efectos anormales. Una definición más satisfactoria es "el estado metabólico en que la respuesta tisular medida en respuesta a la insulina es menor a la esperada para su concentración disponible aparente".8 Lo anterior incluye el gasto metabólico, secreción de neuropéptidos, músculo liso, endotelio, plaquetas y eritrocitos. La función anabólica de la insulina promueve el uso y almacenamiento intracelular de glucosa, aminoácidos y ácidos grasos libres (AGL) e inhibe los procesos catabólicos. En la RI se requiere más insulina para una respuesta normal. Aunque presente en casi todo diabético, existe en individuos que aún no presentan hiperglucemia pero sí SM. Antes de la DM, el paciente sobre–produce insulina para mantener la glucosa normal.54 En un punto ocurre insuficiencia de la célula beta, la insulina cae y la glucosa aumenta; por desgracia, la DM se diagnostica cuando aparece la hiperglucemia. si el paciente con RI presenta una glucemia normal, la insulina en ayuno se eleva. Así, con frecuencia, la RI es el último componente del SM que se diagnostica, aunque es la alteración subyacente esencial.53
Efectos de la insulina sobre las plaquetas
Las plaquetas disponen de un receptor para insulina funcional y capaz de autofosforilarse. La insulina disminuye la respuesta plaquetaria al ADP, colágena, trombina, AA y PAF al disminuir el número de receptores adrenérgicos alfa2. Como la adrenalina potencia los efectos de otros agonistas y estimula la inhibición por adenilatociclasa, la insulina, al modificar la acción de la adrenalina, disminuye la respuesta a otros agonistas. En la DM–2 disminuye el número y la función (afinidad) del receptor plaquetario para insulina, lo que sugiere que la hiposensibilidad plaquetaria a esta hormona contribuye a la hiperactividad de la plaqueta.55 Es factible que la insulina mantenga la sensibilidad a la PGI2, teoría que se fortalece por la falta de respuesta plaquetaria a la PGI2 en la EAC, la cual se normaliza con insulina.31,56 Esto sucede porque la insulina aumenta los sitios de unión a la PGI2 e incrementa la respuesta del cAMP a esta prostaglandina. En las plaquetas de obesos con RI, la insulina altera la inhibición de la agregación en comparación con controles sanos delgados. Por otra parte, la vitamina E y la troglitazona tienen efecto antiplaquetario al suprimir la señal inducida por la trombina que hidroliza PI.57 Dosis entre 200 y 400 mg/día de troglitazona, con o sin insulina, disminuyen la concentración del PAI–1 y mejoran la respuesta fibrinolítica de la DM.57 Para finalizar, la RI reduce la sensibilidad endotelial a la insulina, se produce menos NO y PGI2 y aumenta el PAI–1. La metformina mejora la respuesta endotelial a la insulina.55 Ya que en la DM–2 y en la RI se pierden efectos inhibitorios de la insulina y de la PGI2 y disminuye la respuesta plaquetaria a la insulina, es que se propone a la plaqueta como un blanco más de la RI.31
Conclusión
En la DM–2 se pierden las acciones inhibitorias de la insulina y de la PGI2 sobre la activación plaquetaria, lo que contribuye a la hiperactividad de las plaquetas y a la ateroesclerosis. En la RI existe una respuesta plaquetaria disminuida a la insulina, al NO y a la NOS. Por ello, la plaqueta parece un blanco más de los efectos deletéreos de la RI. En parte, el riesgo de ateroesclerosis prematura y las complicaciones trombóticas propias de la DM–2 se explican por la pérdida de la función vasodilatadora, antitrombótica y antiinflamatoria del endotelio y por hiperactividad plaquetaria. La disfunción plaquetaria de la DM–2 se caracteriza por hiperactividad (adhesión y agregación) de las plaquetas y resistencia de las mismas a la inhibición de la insulina sobre su función. En la DM–2 se altera el metabolismo plaquetario debido a cambios en las vías de señalización intraplaquetaria. La mala regulación de la actividad plaquetaria es clave en la patogénesis de la ateroesclerosis y de las complicaciones vasculares de la DM.
Bibliografía
1. Sistema Nacional de Información en Salud, Secretaría de Salud, Anuario Estadístico 2003. [ Links ]
2. Powers AC. Diabetes mellitus. En: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors: Harrison's principles of internal medicine. México, D. F., Mcgraw–Hill Interamericana 2006:2367–97. [ Links ]
3. Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12–yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16:434–44. [ Links ]
4. Beckman JA, Creager M, Libby P. Diabetes and atherosclerosis. Epidemiology, pathophysiology, and management. JAMA 2002;287:2570–81. [ Links ]
5. Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998;339:229–34. [ Links ]
6. Majluf Cruz A. Fisiología del sistema de coagulación. En: Majluf Cruz A, Pérez Ramírez OJ (eds.): Hematología básica. México, D. F., Editorial garmarte 2006:163–9. [ Links ]
7. Ashby B, Daniel JL, Smith JB. Mechanisms of platelet activation and inhibition. Hematol Oncol Clin North Am 1990;4:1–26. [ Links ]
8. Vinik AI, Erbas T, Park TS, Nolan R, Pittenger GL. Platelet dysfunction in type 2 diabetes. Diabetes Care 2001 ;24:1476–85. [ Links ]
9. Knight DE, Hallam TJ, Scrutton MC. Agonist selectivity and second messenger concentration in Ca2+ –mediated secretion. Nature 1982;296:256–7. [ Links ]
10. Watson SP, Ruggiero M, Abrahams SL. Inositol 1,4,5–trisphosphate induces aggregation and release of 5–hydroxytryptamine from saponin–permeabilized human platelets. J Biol Chem 1986;261:5368–72. [ Links ]
11. Gerrard JM, Carroll RC. stimulation of protein phosphorylation by arachidonic acid and endoperoxide analog. Prostaglandins 1982;22:81–94. [ Links ]
12. FitzGerald GA. Mechanisms of platelet activation: Thromboxane A2 as an amplifying signal for other agonists. Am J Cardiol 1991;68:11B–5. [ Links ]
13. Offermanns S, Laugwitz KL, Spicher K. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc Natl Acad Sci USA 1994;91:504–8. [ Links ]
14. Walker TR, Watson SP. Synergy between Ca2+ and protein kinase C is the major factor in determining the level of secretion from human platelets. Biochem J 1993;289:277–82. [ Links ]
15. Keularts IMLW, van Gorp RMA, Feijge MAH. Alpha2A–adrenergic receptor stimulation potentiates calcium release in platelets by modulating cAMP levels. J Biol Chem 2000;275:1763–72. [ Links ]
16. Brass LF. Ca2+ homeostasis in unstimulated platelets. J Biol Chem 1984;259:12563–70. [ Links ]
17. Clemetson KJ, Clemetson JM. Platelet receptors. En: Michelson AD (ed.): Platelets. London, UK, Elsevier 2007:117–45. [ Links ]
18. Wilde JI, Retzer M, Siess W. ADP–induced platelet shape change: An investigation of the signaling pathways involved and their dependence on the method of platelet preparation. Platelets 2000;11:286–95. [ Links ]
19. Stephens G, Yan Y, Jandrot–Perrus M, Villeval JL, Clemetson KJ, Phillips DR. Platelet activation induces metalloproteinase–dependent GP VI cleavage to down–regulate platelet reactivity to collagen. Blood 2005;105:186–91. [ Links ]
20. Knezevic I, Borg C, Le Breton GC. Identification of Gq as one of the G–proteins which copurify with human platelet thromboxane A2/prostaglandin H2 receptors. J Biol Chem 1993;268:26011–7. [ Links ]
21. Mills DC. ADP receptors on platelets. Thromb Haemost 1996;76:835–56. [ Links ]
22. Packham MA, Mustard JF. Platelet aggregation and adenosine diphosphate/adenosine triphosphate receptors: a historical perspective. Semin Thromb Hemost 2005;31:129–38. [ Links ]
23. Rolf MG, Brearley CA, Mahaut–Smith MP. Platelet shape change evoked by selective activation of P2X1 purinoceptors with alpha, beta–methylene ATP. Thromb Haemost 2001;85:303–8. [ Links ]
24. Leon C, Hechler B, Vial C, Leray C, Cazenave JP, Gachet C. The P2Y1 receptor is an ADP receptor antagonized by ATP and expressed in platelets and megakaryoblastic cells. FEBs Lett 1997;403:26–30. [ Links ]
25. Oury C, Toth–Zsamboki E, Thys C, Tytgat J, Vermylen J, Hoylaerts MF. The ATP–gated P2X1 ion channel acts as a positive regulator of platelet responses to collagen. Thromb Haemost 2001;86:1264–71. [ Links ]
26. Mazzucato M, Cozzi MR, Pradella P, Ruggeri ZM, De Marco L. Distinct roles of ADP receptors in von Willebrand factor–mediated platelet signaling and activation under high flow. Blood 2004;104:3221–7. [ Links ]
27. Boyer JL. Perspective in the use of P2 agonists/antagonists in clinical medicine. Haematologica 2002;87(suppl 1 ):1–30. [ Links ]
28. Savi P, Beauverger P, Labouret C, Delfaud M, Salel V, Kaghad M, et al. Role of P2Y1 purinoceptor in ADP–induced platelet activation. FEBs Lett 1998;422:291–5. [ Links ]
29. Brass LF. The molecular basis for platelet activation. En: Hoffman R (ed.): Hematology. Basic principles and practice. New York, Churchill Livingstone 2000:489–501. [ Links ]
30. Li Y, Woo V, Bose R. Platelet hyperactivity and abnormal Ca2+ homeostasis in diabetes mellitus. Am J Physiol Heart Circ Physiol 2001; 280:H1489. [ Links ]
31. Ferroni P, Basili S, Falco A, Davi G. Platelet activation in type 2 diabetes mellitus. J Thromb Haemost 2004;2:1282–91. [ Links ]
32. Colwell JA, Nesto EW. The platelet in diabetes: focus on prevention of ischemic events. Diabetes Care 2003;26:2181–8. [ Links ]
33. Mammen EF. Sticky platelet syndrome. Semin Thromb Hemost 1999;25:361–5. [ Links ]
34. Davi G, Gresele P, Violi F, Basili S, Catalano M, Giammarresi C, et al. Diabetes mellitus, hypercholesterolemia, and hypertension but no vascular disease per se are associated with persistent platelet activation in vivo. Evidence derived from the study of peripheral arterial disease. Circulation 1997;96:69–75. [ Links ]
35. Noumura S. Platelet activation markers. Rinsho Byori 2003;51:1096–101. [ Links ]
36. Watala C, Golanski J, Pluta J, Boncler M, Rozalski M, Luzak B. Reduced sensitivity of platelets from type 2 diabetic patients acetylsalicylic acid (aspirin)–its relation to metabolic control. Thromb Res 2004; 113:101–13. [ Links ]
37. Shouzu A, Noumura S, Omoto S, Hayakawa T. Effect of ticlopidine on monocyte–derived microparticles and activated platelet markers in diabetes mellitus. Clin Appl Thromb Hemost 2004; 10:167–73. [ Links ]
38. Fattah MA, Shaheen MH, Mahfouz MH. Disturbances of haemostasis in diabetes mellitus. Dis Markers 2003–2004;19:251–8. [ Links ]
39. Bastyr EJ III, Kadrofske MM, Vinik AI. Hyperaggregatory function of platelets in type 1 diabetic subjects (IDDM) occurs in receptor–specifc first phase. Diabetes 1987;36(suppl 1):208A. [ Links ]
40. Gerrard JM, Stuart MJ, Rao GHR, Steffes MW, Mauer SM, Brown DM, et al. Alteration in the balance of prostaglandin and thromboxane synthesis in diabetic rats. J Lab Clin Med 1980;95:950–8. [ Links ]
41. Davi G, Catalano I, Averna M, Notarbartolo A, Strano A, Ciabattoni G, et al. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N Engl J Med 1990;322:1769–74. [ Links ]
42. Bastyr EJ III, Kadrosfske MM, Dershimer RC, Vinik AI. Decreased platelet phosphoinositide turnover and enhanced platelet activation in IDDM. Diabetes 1989;38:1097–102. [ Links ]
43. Paolisso G, Barbagallo M. Hypertension, diabetes mellitus, and insulin resistance: the role of intracellular magnesium. Am J Hyperten 1997;10:346–55. [ Links ]
44. Pellegrata F, Folli F, Ronchi P, Caspani L, Galli L, Vicari AM. Deranged platelet calcium homeostasis in poorly controlled IDDM patients. Diabetes Care 1993;16:178–83. [ Links ]
45. Tschope D, Rosen P, Gries FA. Increase in the cytosolic concentration of calcium in platelet of diabetics type II. Thromb Res 1991;62:421–8. [ Links ]
46. Schechter M, Merz CN, Paul–Labrador MJ, Kaul S. Blood glucose and platelet–dependent thrombosis in patient with coronary artery disease. J Am Coll Cardiol 2000;35:300–7. [ Links ]
47. Bonnefont Rousselot D, Bastard JP, Jaudon MC, Delattre J. Consequences of the diabetic status on the oxidant/antioxidant balance. Diabetes Metab 2000;26:163–76. [ Links ]
48. Tschope D, Diersch E, Scwippert B, Nieuwenhuis HK, Gries FA. Exposure of adhesion molecules on activated platelets in patients with newly diagnosed IDDM is not normalized by near–normoglycaemia. Diabetes 1995;44:890–4. [ Links ]
49. Grundy SM, Brewer B, Cleeman JI, Smith SC, Lenfant C. Definition of metabolic syndrome. Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation 2004;109:433–8. [ Links ]
50. Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care 2003;51:1876–83. [ Links ]
51. Grundy SM. Hypertriglyceridemia, atherogenic dyslipidemia, and the metabolic syndrome. Am J Cardiol 1998;81:18B–25. [ Links ]
52. Nakamura T, Tokunga K, Shimomura I, Nishida M, Yoshida S, Kotani K, et al. Contribution of visceral fat accumulation to the development of coronary artery disease in non–obese men. Atherosclerosis 1994;107:239–46. [ Links ]
53. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: finding from the Third National Health and Nutrition Examination survey. JAMA 2002;287:356–9. [ Links ]
54. Tenenbaum A, Fisman EZ, Motro M, et al. Metabolic syndrome and type 2 diabetes mellitus: focus on peroxisoma proliferator actived receptors (PPAR). Cardiovasc Diabetol 2003;2:4. [ Links ]
55. Udvary M, Pfliegler G, Rak K. Platelet insulin receptor determination in non–insulin dependent diabetes mellitus. Experientia 1985;41:422–3. [ Links ]
56. Kahn NN, Najeeb MA, Ishaq M, Rahim A. Normalization of impaired response of platelet to prostaglandin E1/12 and synthesis of prostacyclin by insulin in unstable angina pectoris and in acute myocardial infarction. Am J Cardiol 1992;70:582–6. [ Links ]
57. Ishizuka T, Itaya S, Wada H, Ishizawa M. Differential effect of the antidiabetic thiazolidinediones troglitazona and pioglitazone on human platelet aggregation mechanism. Diabetes 1998;47:1494–1500. [ Links ]

