










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos de cardiología de México
On-line version ISSN 1665-1731Print version ISSN 1405-9940
Arch. Cardiol. Méx. vol.79 suppl.2 Ciudad de México Dec. 2009
PARTE I
Bases de las arritmias
Genética en los Síndromes de QT prolongado
Genetic in long QT syndromes
Pedro Iturralde–Torres,1 Argelia Medeiros–Domingo.2
1 Subjefe del servicio de Electrofisiología. Instituto Nacional de Cardiología Ignacio Chávez.
2 Médico adjunto en el Sudden Death Genomics Laboratory. Mayo Clinic, Rochester, Minnesota. EUA.
Correspondencia:
Dr. Pedro Iturralde Torres.
Departamento de Electrocardiografía.
Instituto Nacional de Cardiología Ignacio Chávez.
Juan Badiano No. 1 Colonia Sección XVI.
Tlalpan. 14080, México, D. F.
Teléfono: (5255) 5513 3740.
Correo electrónico: pedro@ yahoo.com
Recibido el 26 de agosto de 2009;
Aceptado el 15 de septiembre de 2009.
Resumen
El síndrome de QT largo (SQTL) es un trastorno genético que se caracteriza por prolongación del intervalo QT en el electrocardiograma (ECG) y propensión a la taquicardia ventricular polimorfa "torsades de pointes", la que con frecuencia origina síncope, falla cardiaca o muerte súbita, por lo general en individuos jóvenes y sanos. El síndrome de QT largo es causado por mutaciones de los genes de los canales iónicos de potasio y de sodio o de las proteínas relacionadas con esos canales, lo que produce una sobrecarga positiva de la célula del miocardio con la consecuente prolongación heterogénea de la repolarización en varias capas y regiones del miocardio. Estas condiciones facilitan posdespolarizaciones tempranas y fenómenos de reentrada, que de forma subyacente desarrollan la taquicardia ventricular polimorfa observada en estos pacientes. La historia clínica detallada con respecto a eventos cardiacos en el paciente y miembros de su familia en combinación con la interpretación cuidadosa del ECG de 12 derivaciones (con la medición del intervalo QT en todos los ECG disponibles y la evaluación morfológica de la onda T) son suficientes para diagnosticar el síndrome. El SQTL presenta gran heterogeneidad genética y se han identificado ya más de 500 mutaciones distribuidas hasta ahora en 10 genes: KCNQ–1, HERG, SCN–5A, KCNE–1, KCNE–2, ANKB, KCNJ–2, CACNA–1, CAV–3 y SCN–4B. A pesar de los avances en la materia, aún 25% a 30% de los pacientes permanece sin diagnóstico genético. Los estudios de genética juegan un papel importante y son útiles en casos con resultados no diagnósticos o de ECG limítrofes.
Palabras clave: Síndrome de QT largo; Genética; Tamizaje genético; Diagnóstico prenatal; México.
Abstract
The long QT syndrome (LQTS) is a genetic disorder characterized by prolongation of the QT interval in the electrocardiogram (ECG) and a propensity to "torsades de pointes" ventricular tachycardia frequently leading to syncope, cardiac arrest, or sudden death usually in young otherwise healthy individuals. The long QT syndrome is caused by mutations of predominantly potassium and sodium ion channel genes or channel–related proteins leading to positive overcharge of myocardial cell with consequent heterogeneous prolongation of repolarization in various layers and regions of myocardium. These conditions facilitate the early after–depolarization and reentry phenomena underlying development of polymorphic ventricular tachycardia observed in patients with LQTS. Obtaining detailed patient history regarding cardiac events in the patient and his/her family members combined with careful interpretation of standard 12–lead ECG (with precise measurement of QT interval in all available ECGs and evaluation of T–wave morphology) usually is sufficient to diagnose the syndrome. The LQTS show great genetic heterogeneity and has been identified more than 500 mutations distributed in 10 genes: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1A, CAV3 and SCN4B. Despite advances in the field, 25–30% of patients remain undiagnosed genetic. Genetic testing plays an important role and is particularly useful in cases with nondiagnostic or borderline ECC findings.
Key words: Long QT syndrome; Genetics; Genetic screening; Prenatal diagnosis; Mexico.
Introducción
El síndrome de QT largo (SQTL) se caracteriza por una grave alteración en la repolarización ventricular traducida en el electrocardiograma (ECG) por un alargamiento en el intervalo QT que predispone a arritmias ventriculares malignas –torsade de pointes– y muerte súbita.1,2
La clasificación utilizada en el pasado se basa en la presentación homocigota o heterocigota de la enfermedad, que dan lugar a los síndromes de Jervell–Lange–Nielsen (J–L–N, con sordera) y Romano Ward (sin sordera), respectivamente. De 1995 a 1996 se describieron los tres principales genes vinculados con la enfermedad. Codifican para unidades formadoras del poro de los canales del potasio IKs e IKr y del sodio Nav1,5 que explican aproximadamente 65% de los casos. Si bien en los años subsecuentes se añadieron siete genes más a la lista, éstos explican tan sólo cerca de 5% de los casos. Los canales iónicos son proteínas transmembrana encargadas de transportar iones a través de la membrana celular; los canales implicados en el SQTL son selectivos o especializados en el transporte de un solo ion y dependientes de voltaje, es decir, su activación ocurre a determinado voltaje intracelular. Los fenómenos eléctricos y contráctiles que suceden en el cardiomiocito son controlados por estas estructuras. Los canales iónicos forman complejos macromoleculares; hay una unidad principal formadora del poro del canal y proteínas auxiliares que lo regulan. La afectación en la función de un canal en el SQTL se puede dar en cualquiera de estas dos proteínas, en la principal o en las reguladoras. La afección en la unidad formadora del poro, conocida como alfa, genera los tres subtipos más comunes de SQTL: SQTL–1 (afección en el canal del potasio IKs), SQTL–2 (afección en el canal del potasio IKr) y SQTL–3 (afección en el canal del sodio). Al ser los más frecuentes, disponen de una mejor caracterización clínica y genética.3
Las primeras manifestaciones arrítmicas ocurren durante la adolescencia y en gran medida se desencadenan por un aumento de la actividad simpática. Las mutaciones en los genes que codifican los canales iónicos o las proteínas que controlan dichos canales han surgido como la base del SQTL.
Mientras que el síndrome de Romano–Ward depende de mutaciones que afectan al menos a cinco genes (y todos ellos codifican subunidades de los canales del potasio y sodio cardiacos: KCNQ–1, KCNH–2, SCN–5A, KCNE–1, y KCNE–2), el síndrome de J–LN depende de mutaciones homocigotas o heterocigotas compuestas en uno o dos genes (KCNQ–1 y KCNE–1).4 Todavía se desconoce si la presencia simultánea de un defecto heterocigoto en KCNQ–1 y KCNE–1 resulta en el síndrome de J–LN. El gen ANK–2, que codifica la anquirina B cardiaca, una proteína estructural que fija los canales iónicos a la membrana celular, ha demostrado causar SQTL del subtipo 4. Esta forma de SQTL es de suma rareza y se han documentado pocos portadores de mutaciones en el gen ANK–2.
Puesto que los pacientes con mutaciones ANK–2 pueden mostrar grados variables de disfunción cardiaca, desde bradicardia y arritmia sinusal hasta fibrilación ventricular idiopática y taquicardia ventricular polimorfa catecolaminérgica y puesto que los intervalos largos de QTc no son una característica sistemática en los portadores de la mutación ANK–2, es razonable considerar la disfunción de la anquirina B como una entidad clínica en sí misma y distinta del SQTL clásico.5
En fecha reciente se identificaron cuatro mutaciones nuevas en CAV–3 (que codifica la caveolina 3) en 905 pacientes sin lazos familiares con pacientes con SQTL. Ello indica que esta nueva forma genética de SQTL es de suma rareza. Las caveolas son conocidos microdominios de membrana cuyo componente principal en el músculo estriado es la caveolina 3. Los canales del sodio cardiaco se ubican en las caveolas. El análisis electrofisiológico de la corriente de sodio demostró que mutantes CAV–3 dan lugar a un aumento dos a tres veces superior en la corriente de sodio tardía si se compara con el gen natural CAV–3, y está bien establecido que este mecanismo fisiopatológico causa prolongación del intervalo QT.6
Síndrome de QT largo tipo 10 (SQTL–10)7
Esta variedad se notificó en un caso muy grave, con un QTc > 700 ms, bradicardia fetal y bloqueo auriculoventricular (AV) 2:1. Resulta de mutaciones en el gen SCN–4B localizado en el cromosoma 11 (11q23) que codifica para la subunidad beta–4 del canal del sodio. Se han descrito cuatro subtipos distintos de subunidades que interactúan y regulan las diversas isoformas del canal del sodio, pero sólo el subtipo 4 se relacionado hasta ahora con arritmogénesis. La incidencia de mutaciones en este gen no ha sido explorada, pero se estima <1%.
El SQTL presenta gran heterogeneidad genética y se han identificado ya más de 500 mutaciones distribuidas hasta ahora en 10 genes: KCNQ–1, HERG, SCN–5A, KCNE–1, KCNE–2, ANKB, KCNJ–2, CACNA–1, CAV–3 y SCN–4B. Pese a los avances en la materia, 25% a 30% de los pacientes permanece sin diagnóstico genético. Es una enfermedad de presentación principalmente monogénica; las variedades poligénicas o compuestas suelen dar un fenotipo más grave. La penetrancia –casos que tienen la mutación y el fenotipo– oscila entre 25% y 90%, y con menos frecuencia puede haber variaciones en la expresividad de la enfermedad, diversos fenotipos que puede dar una misma mutación. Los estudios genéticos moleculares desarrollados en los últimos 11 años han permitido realizar una importante correlación entre genotipo y fenotipo y orientar así el tratamiento; también se han hecho interesantes observaciones en cuanto a la susceptibilidad individual para desarrollar arritmias al estudiar los efectos de polimorfismos no equivalentes frecuentes en la población, lo que ha motivado gran interés, sobre todo en el área de la farmacogenómica.
Tamizaje genético
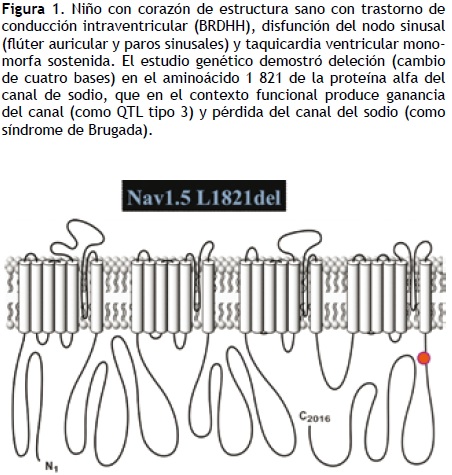
En los últimos años, el estudio genético del SQTL ha permanecido limitado a laboratorios de investigación; sin embargo, la información derivada de éste es de gran utilidad en el tratamiento de los enfermos, en particular en los casos de alto riesgo. La principal aplicación es, quizá, el consejo genético, pero también tiene importantes implicaciones en el tratamiento, que puede orientarse según el canal afectado (Figura 1). La localización precisa de una determinada mutación puede otorgar información adicional en cuanto a la evaluación del riesgo. Las mutaciones en la región transmembrana de KCNQ–1 (IKs) tienen mayor probabilidad de presentar fenómenos arrítmicos que las mutaciones en la región C terminal, así como las mutaciones en la región del poro de KCNH–2 o HERG comparadas con mutaciones en la región N o C terminal. El escrutinio inicial quizá se pueda limitar a los genes KCNQ–1, HERG y SCN–5A, con posibilidad de encontrar mutaciones en 65% de los casos; en caso de obtener resultados negativos, se puede ampliar el tamizaje a los genes KCNE–1, KCNE–2, ANKB, KCNJ–2, CACNA–1, CAV–3 y SCN–4B, lo que permitirá incrementar la posibilidad de resultado positivos en 5 a 10 por ciento.8–10
Polimorfismos reguladores
Se han descrito diversos y frecuentes polimorfismos en la población distribuidos en casi todos los genes asociados al SQTL. Si bien estos cambios no parecen ser patogénicos, algunos pueden tener los siguientes efectos: 1) generar susceptibilidad individual para desarrollar arritmias; 2) favorecer el efecto patogénico de otro cambio no equivalente; 3) disminuir el efecto patogénico de otro cambio no equivalente. Así sucede con el polimorfismo K–897T en KCNH–2 (HERG), con una frecuencia en la población de hasta 15%, que no sólo se ha asociado con susceptibilidad a determinados fármacos, sino que también favorece el efecto patogénico de mutaciones en el mismo gen. Otro ejemplo es el polimorfismo S–1103Y en el gen SCN–5A, reconocido sobre todo en la población de raza negra, con una incidencia cercana a 13% y asociado con un incremento en el riesgo de muerte súbita infantil. Interesante también ha sido la descripción de dos sitios de procesamiento alternativo en el producto del gen SCN–5A, que codifica la isoforma del canal del sodio cardiaco Nav–1,5 en el ser humano, puesto que genera dos tipos de canales del sodio: uno con 2 016 aminoácidos que contiene una glutamina en la posición 1 077 (Q–1077) y otro con 2 015 aminoácidos que carece de esta glutamina (Q–1077del). Transcritos de estos dos procesamientos alternativos están presentes en un mismo corazón humano a razón de 2:1 y diversos polimorfismos frecuentes tendrán un efecto distinto en la función del canal dependiendo del contexto Q–1077 o Q–1077del. De manera inicial, lo anterior se demostró con el polimorfismo H–558R de SCN–5A, presente hasta en 30% de la población; al expresar H–558R en el contexto de Q–1077, se observó una profunda disminución de la corriente iónica. Un efecto similar se documentó con el polimorfismo S–524Y. Estos hallazgos han suministrado elementos para explicar la gravedad variable de la enfermedad, así como los distintos fenotipos de una misma mutación observados en algunas familias.11
Tamizaje genético post mortem
Interesante ha sido el hallazgo de mutaciones en genes que condicionan SQTL en casos de niños con muerte súbita infantil y en casos de muerte súbita inexplicable en el adulto joven. El estudio genético post mortem en pacientes con muerte súbita y autopsia negativa ha mostrado mutaciones que condicionan SQTL en porcentajes variables cercanos a 10% en la muerte súbita infantil y a 35% en la muerte súbita del adulto joven. Sobre la base de estos resultados, se ha propuesto realizar un ECG sistemático en todos los neonatos. El estudio genético post mortem, también conocido en la literatura científica como "autopsia molecular", además de tener repercusiones legales, tiene importantes implicaciones en los familiares de los casos que pudieran estar afectados sin saberlo.
Hallazgos genéticos en el síndrome de muerte súbita infantil y en el síndrome de muerte súbita inexplicada
Un estudio molecular con 201 víctimas del síndrome de muerte súbita infantil (SMSI) en Noruega, reveló que las variantes genéticas en los genes del SQTL están presentes en 9.5% de las víctimas de SMSI. Un estudio muy reciente documentó las mutaciones CAV–3 como una de las bases patogénicas del SMSI: se identificaron tres mutaciones CAV–3 diferenciadas en tres de 50 lactantes de raza negra, pero no se detectó ninguna mutación CAV–3 en 83 lactantes de raza blanca. Dada la creciente coherencia entre éstos y otros informes genéticos post mortem, el reto es encontrar un modo eficaz y asequible de detectar el SQTL presintomático para reducir la morbilidad y la mortalidad en el subconjunto de muertes súbitas infantiles etiológicamente relacionadas con los genes del SQTL, así como determinar si es apropiado establecer criterios electrocardiográficos en los recién nacidos. En otro minucioso y reciente estudio, se realizaron pruebas genéticas post mortem en 49 víctimas de muerte súbita inexplicable cuyo fallecimiento había tenido lugar a una edad promedio de 14.2 años. Más de un tercio de los fallecidos portaba una supuesta mutación en los canales cardiacos: siete portaban mutaciones en el canal de liberación del calcio codificado por RyR–2 y otros 10 de ellos mutaciones que suscitaban propensión al SQTL. En consecuencia, si la evaluación post mortem de la causa de muerte súbita resulta negativa, es aconsejable realizar pruebas genéticas post mortem de los canales cardiacos para ofrecer consejo genético fiable a los familiares de la víctima.
Subgrupos específicos
Adolescentes. Puesto que en el análisis de los predictores de sucesos cardiacos el síncope se considera un criterio de valoración crucial, se realizó un estudio específico con 2 772 pacientes con SQTL para identificar los factores de riesgo asociados con paro cardiaco controlado y muerte cardiaca súbita durante la adolescencia. Las variables predictivas independientes y significativas de paro cardiaco controlado o de muerte cardiaca súbita durante la adolescencia fueron el síncope, el intervalo QTc y el sexo. Los pacientes con un episodio de síncope en los últimos dos años presentaban un cociente de riesgo instantáneo ajustado (hazard ratio [HR]) de 11.7 (intervalo de confianza [IC] al 95%, 7.0–19.5; p < 0.001), y los que tenían dos o más episodios de síncope en los últimos dos años presentaban un HR ajustado de 18.1 (IC al 95%, 10.4–31.2; p < 0.001) de casos potencialmente mortales. Con independencia de los sucesos ocurridos más de dos años atrás, un QTc ≥ 530 ms se asoció con mayor riesgo (HR ajustado = 2.3; IC al 95%, 1.6–3.3; p < 0.001) respecto a un QTc inferior. Los varones con edades comprendidas entre los 10 y los 12 años presentaban mayor riesgo que las mujeres (HR = 4.0; IC al 95%, 1.8–9.2; p = 0.001), pero no había diferencias de riesgo significativas entre ambos sexos entre las edades de 13 y 20 años.12
Diagnóstico prenatal de síndrome de QT largo
La bradicardia fetal puede ser una de las primeras manifestaciones clínicas del SQTL. En series retrospectivas se ha documentado que hasta 70% de los pacientes diagnosticados en la infancia tiene este antecedente, que suele ir acompañado de hidropesía fetal. La evaluación de la repolarización cardiaca fetal entre las semanas 14 y 39 es un método útil para el diagnóstico oportuno del SQTL. Mosaicismos gonadales para SQTL se han asociado con pérdidas fetales recurrentes durante el tercer trimestre del embarazo. Si la sospecha de la enfermedad es muy alta, la amniocentesis a partir de las 16 semanas de gestación puede ser de utilidad para el diagnóstico, que resulta sencillo cuando alguno de los progenitores se conoce como portador de una mutación determinada.
La arritmia ventricular característica del SQTL es la conocida "torsade de pointes". Se presenta cuando el intervalo QT se prolonga, independientemente de la etiología. Es una taquicardia ventricular polimorfa por reentrada que en el electrocardiograma se caracteriza por un giro continuo del eje del QRS sobre una línea imaginaria. Suele estar precedida por una pausa seguida de una extrasístole (intervalo RR "corto–largo–corto"), y puede terminar en fibrilación ventricular y muerte súbita. Si esto no sucede, el paciente puede experimentar sólo un síncope o incluso, si el episodio es breve, puede pasar desapercibido.
Ubicación de la mutación en el canal
Según un estudio reciente, los pacientes con QTL–2 y mutaciones en la región del poro del gen KCNH–2 presentan un riesgo bastante mayor de experimentar sucesos cardiacos relacionados con arritmias que las personas con mutaciones en otras regiones. Además de las mutaciones en el poro, aquéllas en el dominio PAS del canal KCNH–2 también pueden conllevar efectos perjudiciales. De igual manera, las mutaciones asociadas con QTL–1 y que implican a los dominios transmembrana o a regiones del poro del canal KCNQ–1 fueron seguidas de un desenlace clínico peor que las mutaciones en el carbono terminal portadas por otros pacientes con QTL–1.
Dado el papel comprobado que la identificación del sustrato genético desempeña en el tratamiento de pacientes con SQTL, el reto más importante en este campo es ahora aumentar el acceso y el reembolso de los análisis genéticos para el SQTL.13
Bibliografía
1. Medeiros DA, Iturralde TP, Ackerman MS. Clínica y genética en el síndrome de QT largo. Rev Esp Cardiol 2007;60(7):739–52. [ Links ]
2. Wilde AA, Bezzina CP. Genetics of cardiac arrhythmias. Heart 2005;91:1352–8. [ Links ]
3. Robert R. Genomics and cardiac arrhythmias. J Am Coll Cardiol 2006;47:9–21. [ Links ]
4. Márquez MF, Ramos KM, Hernández PG, Estrada J, Frebegat JR, Pérez VN, et al. Detección de una mutación en el gen KCNQ–1 (KvLQT–1) causante de síndrome de Jervell–Lange–Nielsen en una familia mexicana. Arch Cardiol Mex 2006;76:3 263–8. [ Links ]
5. Marban E. Cardiac channolopathies. Nature 2002;415:213–8. [ Links ]
6. Priori SG, Napolitano C. Role of genetic analysis in cardiology. Part I. Mendelian disease. Cardiac channelopathies. Circulation 2006;113:1130–5. [ Links ]
7. Medeiros DA, Kaku T, Tester DJ, Iturralde TP, Itty P, Ye B, et al. SCN4B– encoded sodium channel beta subunit in congenital long QT syndrome. Circulation 2007; 116:134–42. [ Links ]
8. Roden DN. Clinical practice. Long QT syndrome. N Engl Med 2008;358:169–76. [ Links ]
9. Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol 2008;51:2291–300. [ Links ]
10. Modell SM, Lehmann MH. The long QT syndrome family of cardiac ion channelopathies: a HuGE review. Genet Med 2006;8(3):143–55. [ Links ]
11. Antzelevitch C. Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. J Cardiovas Electrophysiol 2003;14:1259–72. [ Links ]
12. Rossenbacker T, Priori SG: Nuevas perspectivas en el síndrome de QT largo. Rev Esp Cardiol 2007;60:675–82. [ Links ]
13. Campuzano O, Sarquella Brugada G, Brugada R, Brugada P, Brugada J. Bases genéticas de las arritmias malignas y las miocardiopatías. Rev Esp Cardiol 2009;62(4):422–36. [ Links ]

