










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
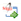 Cited by SciELO
Cited by SciELO Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos de cardiología de México
On-line version ISSN 1665-1731Print version ISSN 1405-9940
Arch. Cardiol. Méx. vol.79 suppl.2 Ciudad de México Dec. 2009
PARTE I
Bases de las arritmias
Avances recientes en la fisiopatología de la fibrilación auricular
Recent advances in the pathophysicology of atrial fibrillation
Manlio F. Márquez,1 Jorge Gómez–Flores,1 Alberto Aranda–Faustro,2 Iris Cazares–Campos,3 Manuel Cárdenas.1
1 Médico adjunto del Departamento de Electrofisiología. Instituto Nacional de Cardiología Ignacio Chávez.
2 Médico adjunto del Departamento de Anatomía Patológica. Instituto Nacional de Cardiología Ignacio Chávez.
3 Pasante de Servicio Social del Departamento de Electrofisiología. Instituto Nacional de Cardiología Ignacio Chávez.
Correspondencia:
Dr. Manlio F. Márquez.
Instituto Nacional de Cardiología Ignacio Chávez.
Departamento de Electrofisiología. Juan Badiano No.1
Col. Sección XVI. Tlalpan, 14080. México, D. F.
Teléfono: (52) 55 5513 3740.
Correo electrónico: manliomarquez@ yahoo.com
Recibido el 24 de agosto de 2009;
Aceptado el 15 de septiembre de 2009.
Resumen
La presente revisión es un resumen de la fisiopatología de la fibrilación auricular (FA) y muestra los avances en el conocimiento de esta arritmia. En su génesis y mantenimiento deben considerarse los factores que se describen a continuación. Factor genético: está implicado en los casos de FA familiar. Factor predisponente estructural: dilatación auricular, el factor estructural más conocido que permite el desarrollo de la FA. Factor predisponente estructural: el haz de Bachmann y las vías de conducción interauricular establecen gradientes de frecuencia entre la aurícula izquierda y la derecha, lo que les confiere un papel en la conducción fibrilatoria. Factores predisponentes electrofisiológicos: periodos refractarios heterogéneos favorecen la generación de FA. Factores desencadenantes: papel de la actividad eléctrica anormal ("focos ectópicos"). Factores moduladores: papel del sistema nervioso autónomo. El aumento del tono vagal acorta los periodos refractarios auriculares, lo que favorece una mayor dispersión de los mismos y la generación de las reentradas en el contexto de una actividad desencadenada. Finalmente, existen tres tipos de remodelamiento auricular secundario a la FA: estructural, contráctil, eléctrico. Los tres se encuentran interrelacionados y contribuyen a mantener la FA; de ahí la sentencia de que "la FA genera más FA".
Palabras clave: Fibrilación auricular; Fisiopatología; Factores predisponentes; Factores desencadenantes; Mantenimiento; Remodelamiento auricular; México.
Abstract
This review is a summary of the pathophysiology of atrial fibrillation (AF) and the progress in the understanding of this arrhythmia. The following factors should be considered in the genesis and maintenance of AF. The genetic factor is involved in cases of familial AF. Predisposing structural factors: atrial dilation, structural feature that allows the development of AF. Predisposing structural factors: the role of the Bachmann s bundle and frequency gradients between the left and right atrium. Electrophysiological factors: heterogeneous refractory periods favor the fibrillatory conduction. Triggers: abnormal electrical activity (ectopic foci). Modulating factors: autonomic nervous system. Increased vagal tone shortens atrial refractory periods, creating a greater dispersion of the refractory periods and the generation of reentries in the context of triggered activity. Finally, there are three types of atrial remodeling secondary to AF: structural, contractile, electrical. They are interrelated and contribute to maintaining the AF ("AF begets AF").
Key words: Atrial fibrillation; Pathophysiology; Predisposing factors; Vagal tone; Maintenance; Atrial remodeling; Atrial refractory period; Mexico.
Introducción
A casi cien años de la primera descripción de la fibrilación auricular (FA) en el ser humano, aún existen muchas interrogantes sobre esta arritmia. Los cardiólogos se preguntan: ¿por qué aumentó la incidencia de FA?, ¿por qué razón ciertas enfermedades no cardiacas se asocian con FA?, ¿existen factores genéticos implicados en la aparición de FA?, ¿qué papel juega la inflamación?, ¿por qué se reconoce ahora que la "FA genera FA"?, ¿es posible prevenir la FA?, ¿cómo actúan los IECA para prevenir la FA?, ¿cuáles son las bases que fundamentan la posibilidad de curar la FA mediante cirugía o ablación con catéter?. La presente revisión es un resumen de la fisiopatología y muestra los avances en el conocimiento de esta arritmia, de la que aún faltan muchas cosas por descubrir.
La FA es una arritmia que se caracteriza por la contracción rápida (entre 400 y 700 por minuto) e irregular de las aurículas. La frecuencia ventricular depende de la capacidad de conducción de los impulsos a través del nodo auriculoventricular (AV) y en forma característica es irregular ("arritmia completa"). Los fisiólogos fueron los primeros en conocer la FA, quienes de manera experimental la inducían mediante estimulación eléctrica de las aurículas. La primera publicación de FA en el ser humano fue de Cushny y Edmonds en 1906.1 Sin embargo, Sir James Mackenzie ya había observado pacientes con FA desde 1880.2 Su observación fue clínica y con registros flebográficos antes de la aparición del ECG; eran enfermos cuya "irregularidad del pulso" se asociaba con ausencia de ondas "a" en los registros del pulso hepático y yugular, por lo que supuso que se trataba de una parálisis auricular. En 1898 encontró en la autopsia de uno de estos enfermos una aurícula distendida y de pared adelgazada, lo que parecía corroborar su hipótesis: la "asistolia auricular". No obstante, en casos subsecuentes encontró lo contrario, es decir, aurículas hipertróficas, evidencia de que estas aurículas se habían estado contrayendo, aunque él no podía registrar dicha actividad. Entonces pensó que la actividad auricular estaba oculta, enmascarada por la actividad ventricular, y que ambas contracciones, auricular y ventricular, se realizaban al mismo tiempo. Supuso que ello se debía a que el impulso eléctrico se originaba en el nodo AV y por ello lo catalogó como "ritmo nodal". Fue hasta que Mackenzie conoció el trabajo de Cushny y Edmonds, quienes descubrieron la FA en el ser humano, cuando reconoció la posibilidad de que en realidad sus pacientes fueran casos de FA. Por su parte, Thomas Lewis, el discípulo favorito de Mackenzie, quien también había inducido FA en perros y conocía lo descrito por Mackenzie, hizo registros electrocardiográficos de la FA experimental en 1909, lo que le permitió correlacionar las oscilaciones de la línea basal del ECG con la FA en el ser humano.
Fisiopatología
En la génesis y mantenimiento de la FA deben considerarse los siguientes factores:
• Factor predisponente genético.
Un factor genético está implicado en los casos de FA familiar.3 Esta entidad fue descrita por primera vez en 1943 por Wolff. Estudios recientes informan una prevalencia de FA familiar correspondiente a 5% de todos los casos de FA y hasta de 15% de todos los casos de FA aislada idiopática. En 1996 Ramón Brugada y colaboradores fueron los primeros en señalar la existencia de una mutación en el locus 10q22–24 (brazo largo del cromosoma 10) en tres familias catalanas afectadas por FA con un modo de herencia autosómico dominante.4 Aunque eran varios los genes candidatos a encontrarse afectados por dicha mutación, fue en el 2003 cuando se informó una mutación (locus 11p15.5) en el gen del canal de potasio KCNQ1 (KvLQT1 ).5 Este gen codifica la formación de los canales de potasio que generan la corriente IKs y la mutación afecta la región del poro del canal, lo que resulta (a diferencia de lo que ocurre en el síndrome de QT largo congénito) en una ganancia de la función. Al incrementarse la salida de K+, la repolarización se hace más precoz, con lo que disminuye la duración de los potenciales de acción que, de producirse de manera no homogénea, provocan la heterogeneidad de los periodos refractarios auriculares implicada en la patogénesis de la FA. En 2003 fue informado también un tercer locus (6q14–16) en una familia de 34 miembros en que ocho sufrían FA. En estos casos se desconoce el gen afectado.6 Como candidatos se han propuesto el gen que codifica el polipéptido alfa de la hormona estimulante de la tiroides (TSH) y el gen de la anquirina. La investigación de estos posibles responsables de la FAes intensa como lo indica en 2004 el grupo de Ramón Brugada, que tenía identificadas seis familias, con 132 individuos, 50 de los cuales tenían FA, dos de ellos diagnosticados in utero.7,8
Hay investigaciones en curso para saber si existe una predisposición genética a adquirir la FA. Yamashita y colaboradores5 compararon la presencia de polimorfismo del gen de la ECA en pacientes con FA idiopática y en sujetos controles.9 Estos polimorfismos, que en el ventrículo están implicados en la generación de fibrosis miocárdica, no son más frecuentes en sujetos con FA. Lai y colaboradores encontraron polimorfismos del alelo 38G del gen minK, aunque se desconoce si este polimorfismo tiene expresión funcional.10
• Factor predisponente estructural: dilatación auricular.
El factor estructural más conocido como sustrato anatómico que permite el desarrollo de la FA es la dilatación de las aurículas. El mayor tamaño auricular y la dilatación auricular producen acortamiento del potencial de acción por disminución de las corrientes Ito e Ical, reducen la fase 0 de despolarización, aumentan el automatismo ectópico e inducen postpotenciales.
La dilatación se acompaña de fibrosis intersticial, la que separa los haces de miocitos, con lo que aumenta la conducción anisotrópica y disminuye la velocidad de conducción, situación que favorece la existencia de FA por la posibilidad de que se formen más circuitos de reentrada en las aurículas.11,12 La dilatación auricular puede ser secundaria a una cardiopatía o a una neumopatía. En el primer caso es importante destacar el papel de la hipertensión arterial sistémica, que al producir disfunción diastólica es capaz de incrementar el tamaño auricular, lo que explicaría en parte la alta prevalencia de FA en este grupo poblacional.13,14 En el segundo caso hay que destacar la asociación entre síndrome de apnea obstructiva del sueño y FA.15 En este caso, el mecanismo involucrado puede consistir en una dilatación de la aurícula derecha secundaria al incremento de la presión arterial pulmonar que puede desarrollarse en estos enfermos.
• Factor predisponente estructural: papel del haz de Bachmann.
En forma experimental, la inducción de FA por estimulación vagal, que inhibe la activación de la ciclasa de adenilato con disminución de la adenosina y del monofosfato cíclico; el aumento que éste produce en Ical disminuye el periodo refractario.
La aparición de focos ectópicos tiene tres patrones que se denominan de reentrada, focal y septal. En el inicio del tipo "focal" la mayor actividad ectópica aparece en la cara anterior de la vena cava superior, en la zona del haz de Bachmann, y tiene una amplia región de activación precoz.16 Otras zonas de actividad focal también aparecen en las orejuelas, cerca de las venas pulmonares derechas y en la aurícula izquierda. También se ha estudiado la forma en que las vías de conducción interauricular (haz de Bachmann y fascículo interauricular inferoposterior) establecen gradientes de frecuencia entre la aurícula izquierda y la derecha, lo que les confiere un papel en la conducción fibrilatoria. Estudios de mapeo óptico en corazón aislado de oveja han demostrado que existe un gradiente de frecuencia de activación progresiva entre la aurícula izquierda y la derecha en la FA.17 Así, una frecuencia dominante en la aurícula izquierda de 18.8 Hz llega a la porción izquierda del haz de Bachmann con una frecuencia de 18.7, mientras que en la porción derecha tiene un frecuencia menor (14.5 Hz). Un gradiente similar, aunque de menor magnitud, se demuestra en la vía interauricular inferoposterior, donde la frecuencia pasa de 14.8 en el lado izquierdo a 14.1 en el lado derecho. Así, al final, la frecuencia promedio en la aurícula derecha es de 9.8 Hz. De estos estudios se deduce la importancia que tiene el haz de Bachmann en la conducción fibrilatoria de la aurícula izquierda a la derecha. Estos hallazgos explicarían en parte la observación hecha en el laboratorio de electrofisiología clínica de actividad fibrilatoria más rápida en la aurícula izquierda que en la derecha.
• Factores predisponentes electrofisiológicos.
Desde principios del siglo pasado surgieron las primeras hipótesis acerca de la generación de FA: foco ectópico, circuito único de reentrada y circuitos múltiples de reentrada. De manera curiosa, los defensores de cada una de estas teorías las consideraban mutuamente excluyentes. Sin embargo, hoy en día se ha demostrado que todos estos mecanismos (reentrada de múltiples ondas de activación ["ondillas"], por un circuito de reentrada errante que cambia de sitio y localización en forma constante ["circuito líder"], por un foco ectópico de descarga muy rápida, por dos focos ectópicos que laten a frecuencias distintas) pueden coincidir, no sólo en un contexto experimental sino también en el ser humano (Figura 1). En cualquier caso, la presencia de periodos refractarios heterogéneos propicia la generación de FA al favorecer bloqueos de conducción que en forma constante cambian de localización y tamaño y que son los que permiten la reentrada que perpetúa la arritmia.
Desde hace varios años el grupo de Jalife18 se halla estudiando los mecanismos de la FA. Al principio realizó simulaciones en computadora para estudiar la propagación de las ondas del circuito en dos dimensiones. Observó que, a diferencia de la teoría del "circuito líder" propuesto en modelos unidimensionales, en su modelo bidimensional las ondas de propagación tenían forma de espiral con un centro excitable ("rotor"). La persistencia y propagación de la espiral de activación dependen del ángulo de curvatura de la misma y de la excitabilidad del tejido auricular. A partir de estos nuevos conocimientos surgieron estudios sobre la propagación del frente de activación en tres dimensiones. Se ha comunicado que se producen ondas en forma de "rollos". Este conocimiento se está aplicando en la terapéutica, ya que se estudia la presencia de rotores en seres humanos con FA e incluso se realiza la ablación con catéter de los mismos para curar la FA19 (Figura 2).

Es interesante señalar la conocida relación entre la aparición de FA en pacientes con vías accesorias auriculoventriculares y el síndrome de Wolff–Parkinson–White (WPW).20 Esto se debe a que, por conducción retrógrada a través del haz anómalo o el nodo, el estímulo llega a la aurícula en el periodo vulnerable y la hace fibrilar. La fibrilación se autosostiene y la activación ventricular se produce unas veces por el haz anómalo y otras veces por el nodo A–V, de acuerdo con los periodos refractarios de la conducción oculta (Figura 3).

Esta hipótesis se ha comprobado, ya que la FA no recurre cuando se practica una ablación exitosa de las vías anómalas. Hace poco se señaló la presencia de dobles potenciales en el seno coronario de pacientes con síndrome de WPW y vulnerabilidad auricular.21
• Factores desencadenantes: papel de la actividad eléctrica anormal ("focos ectópicos").
Pueden ser el "gatillo o disparador" de una FA (Figura 4). Desde hace tiempo se conoce el papel de las extrasístoles en la FA. Mackenzie escribió en 1914: "...he estudiado cientos de casos y visto iniciar esta condición bajo una variedad de circunstancias, en particular en individuos con extrasístoles frecuentes". En 1931 Schwarz publica un artículo titulado "los efectos de la digital sobre las contracciones auriculares prematuras asociadas con ataques de FA paroxística" y en 1932 Langeron se refiere a la "existencia de un estado prefibrilatorio". En 1958 Scherf y colaboradores establecen que un foco ectópico podría generar FA debido a una descarga muy rápida del mismo que no permite la conducción hacia las aurículas en relación 1:1, de lo que resulta FA. Se conocen tres mecanismos por los que se puede originar un foco ectópico: aumento del automatismo, postdespolarizaciones tardías (y actividad desencadenada) y reentrada. En forma experimental se puede inducir FA por una estimulación auricular rápida. Existen múltiples zonas generadoras de actividad ectópica que puede resultar en FA: aurícula derecha (incluida la cresta terminal), aurícula izquierda y el denominado "sistema venoso" (desembocadura de venas pulmonares, vena cava superior, vena cava inferior, ligamento de Marshall).22–24

Apenas en la década pasada Haïssaguerre y su grupo establecieron que la presencia de actividad eléctrica en las venas pulmonares (VP) se asociaba con la génesis y el mantenimiento de algunas formas de FA idiopática25 (Figura 5). Ello constituyó un avance fundamental en el tratamiento de la FA debido a que permitió el desarrollo de la ablación con catéter para el tratamiento de esta arritmia.26 Aunque ya se conocía la existencia de músculo dentro de las VP, fue este descubrimiento el que atrajo la atención de los electrofisiólogos básicos y clínicos hacia estas estructuras. Los avances han sido enormes. Así, el origen de esta actividad se explica en la capacidad del músculo que rodea la porción proximal de las VP para generar potenciales de acción debido a corrientes iónicas activadas por estiramiento al paso del flujo sanguíneo.27 Se ha descrito la presencia de conducción decremental y de periodos refractarios cortos en algunas VP "arritmógenas", lo que sugiere un mecanismo de reentrada en la génesis de esta actividad eléctrica ectópica.28–30Incluso se ha identificado la presencia de células especializadas en conducción (células P, transicionales y de Purkinje) dentro de las VP humanas.31

• Factores moduladores: papel del sistema nervioso autónomo.
El remodelamiento anatómico se caracteriza por una distribución despareja de las terminaciones nerviosas y de los receptores simpáticos y parasimpáticos en las aurículas, lo que acorta el periodo refractario auricular en forma heterogénea (Figura 6).
La acetilcolina actúa por medio de receptores muscarínicos que a través de la proteína G1 inhiben Ical y en cambio activan Ikach, por lo que se acorta la duración del periodo refractario auricular de manera heterogénea y se disminuye la longitud de onda, lo que produce una fibrilación auricular estable.32 En términos generales, se atribuye un mayor componente simpático a la FA relacionada con una cardiopatía estructural y un gran componente vagal a la FA idiopática conocida también como "FA vagal", que describió de manera original Coumel. La asociación entre esofagitis por reflujo y FA puede tener un componente vagal de fondo.33
Por otro lado, se ha informado que alrededor de 70% de los episodios de FA ocurre durante el día y es precedido por un incremento del tono simpático. El 30% de los episodios que ocurren por la noche se relaciona a su vez con un incremento de la actividad vagal. Recientemente Bettoni y colaboradores34exploraron la variabilidad de la frecuencia cardiaca previa al paroxismo de FA relacionada con actividad ectópica en las VP. Encontraron un incremento del intervalo RR promedio y de la banda de frecuencia baja que ocurría hasta 15 minutos antes de los episodios, lo que traduce un incremento del tono simpático, con posterior disminución a los 10 y 5 minutos previos a los episodios en relación con un "retiro simpático", mientras que la banda de frecuencia alta se mantenía elevada (tono parasimpático elevado).35
Un incremento del tono vagal acorta los periodos refractarios auriculares, favorece una mayor dispersión de los mismos y la generación de las reentradas en el contexto de actividad desencadenada presente. Estos conocimientos han tenido aplicación terapéutica. Pappone ha informado la utilidad de la ablación con catéter de la inervación vagal de la aurícula izquierda en la ablación con catéter de la FA36 (Figura 7). La mayor zona de inervación vagal se localiza en la pared posterior de la aurícula izquierda, lo que podría explicar el beneficio de las líneas de ablación realizadas para la "modificación del sustrato" en la FA.37
• Factores de mantenimiento, fenómeno de remodelamiento.
Se denomina remodelamiento a la adaptación fisiopatológica de las aurículas al ritmo fibrilatorio. Existen tres tipos de remodelamiento auricular secundario a la FA: estructural, contráctil y eléctrico. Los tres se encuentran interrelacionados y contribuyen a mantener la FA; de allí la sentencia coloquial de que la "FA genera más FA".
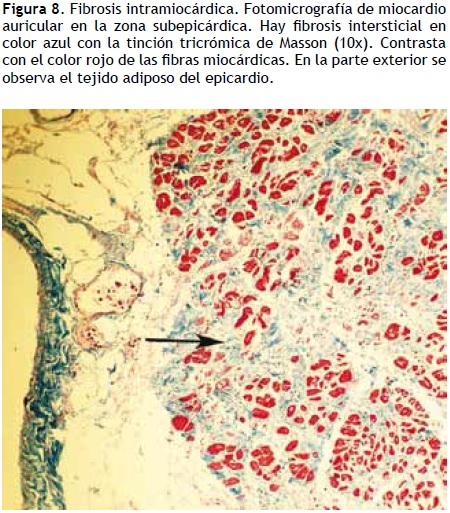
Remodelamiento estructural: se observan varios cambios histopatológicos en las aurículas de individuos con FA, incluidos aquéllos con la denominada FA idiopática.38 Los más distintivos incluyen la presencia de inflamación, la destrucción de fibras musculares (miólisis), el incremento de fibras de colágena en la matriz extracelular y el almacenamiento de glucógeno intracelular (Figura 8). En este sentido, es interesante la observación clínica de Chung y colaboradoress39 de que los pacientes con FA presentan niveles de proteína C reactiva (PCR) mayores que los controles y de que hay una mayor elevación de PCR a mayor "carga" de FA. A los cambios propios de la FA se deben agregar aquellos producidos por la ablación con catéter (Figura 9).

Remodelamiento contráctil: aturdimiento auricular. La FA condiciona disminución de la fuerza de contracción auricular, aun con periodos cortos de taquicardia. Cuando la arritmia termina, esta disminución de la fuerza contráctil se hace más evidente y constituye el denominado "aturdimiento" auricular.40 Este hecho favorece la estasis sanguínea (con su correlación clínica en la presencia de "contraste espontáneo" en los ecocardiogramas de estos pacientes) y a este fenómeno se atribuyen en gran parte los episodios tromboembólicos observados en la FA, aunque la contribución de alteraciones en la coagulación también son muy importantes.41 Remodelamiento eléctrico. La FA sostenida condiciona alteraciones en las corrientes iónicas de los miocitos auriculares que a la larga condicionan mayor dispersión de los periodos refractarios, lo que favorece el mantenimiento de la FA. El importante incremento de la frecuencia de contracción auricular condiciona una sobrecarga de calcio intracelular que amenaza la viabilidad de la célula. Por ello, se inactivan canales del calcio y a largo plazo pueden ocurrir alteraciones moleculares (disminución de RNA mensajero para formar canales de calcio) que contribuyen aún más a disminuir la corriente de calcio. Esto condiciona acortamiento de la duración del potencial de acción, lo que conlleva a la dispersión de los periodos refractarios antes descrita. Uno de los canales más comprometidos en este fenómeno es el de la corriente lenta de calcio (ICaL) por dos mecanismos: regulación a la baja de subunidades del canal y disfunción del canal por cambios en el estado de fosforilación.
Una observación importante es que el remodelamiento eléctrico ocurre en forma concomitante con el remodelamiento contráctil, como lo demostró Schotten.42 En animales de experimentación constató una disminución paralela del índice de trabajo miocárdico auricular y de los periodos refractarios auriculares tras el establecimiento de la FA, con una recuperación igual de paralela tras el restablecimiento del ritmo sinusal
Conclusiones
La FA es una arritmia con un solo cuadro clínico y electrocardiográfico, pero con múltiples mecanismos participantes en su génesis y mantenimiento. Todos los mecanismos descritos en forma experimental desde principios del siglo pasado se han demostrado en el humano. El conocimiento de los focos ectópicos auriculares, del papel del sistema nervioso autónomo, así como de la existencia de rotores está contribuyendo en forma reciente a los avances en la ablación con catéter y al desarrollo de nuevos fármacos para prevenir y tratar esta arritmia.
Bibliografía
1. Cárdenas M. La fibrilación auricular. PAC–EFC–1. México, Intersistemas 1998. [ Links ]
2. Mackenzie J. Diseases of the heart. 3a ed. 1914:211–6. Tomado de: Willius FA, Keys TE. Classics of Cardiology, Vol. 2. Nueva York, Henry Schuman Inc. 1941:769–800. [ Links ]
3. Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 1997;96(4):1180–4. [ Links ]
4. Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, Mont L, et al. Identification of a genetic locus for familial atrial fibrillation. N Engl JMed 1997;336:905–11. [ Links ]
5. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain–of–function mutation in familial atrial fibrillation. Science 2003;299:251–4. [ Links ]
6. Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, et al. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol 2003;41 (12):2185–92. [ Links ]
7. Hong K, Piper DR, Diaz–Valdecantos A, Brugada J, Oliva A, Burashnikov E, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res 2005;68:433–40. [ Links ]
8. Brugada R. Is atrial fibrillation a genetic disease? J Cardiovasc Electrophysiol 2005;16:553–6. [ Links ]
9. Yamashita T, Hayami N, Ajiki K, Oikawa N, Sezaki K, Inoue M, et al. Is ACE gene polymorphism associated with lone atrial fibrillation? Jpn Heart J 1997;38(5):637–41. [ Links ]
10. Lai LP, Su MJ, Yeh HM, Lin JL, Chiang FT, Hwang JJ, et al. Association of the human minK gene 38G allele with atrial fibrillation: evidence of possible genetic control on the pathogenesis of atrial fibrillation. Am Heart J 2002;144(3):485–90. [ Links ]
11. Bolca O, Akdemir O, Eren M, Dagdeviren B, Yildirim A, Tezel T. Left atrial maximum volume is a recurrence predictor in lone atrial fibrillation: an acoustic quantification study. Jpn Heart J 2002;43(3):241–8. [ Links ]
12. Efremidis M, Sideris A, Prappa E, Fillipatos G, Athanasias D, Kardara D, et al. Effect of atrial pressure increase on effective refractory period and vulnerability to atrial fibrillation in patients with lone atrial fibrillation. J Interv Card Electrophysiol 1999;3(4):307–10. [ Links ]
13. Reiffel JA. Is arterial stiffness a contributing factor to atrial fibrillation in patients with hypertension? A preliminary investigation. Am J Hypertens 2004;17(3):213–6. [ Links ]
14. Jais P, Peng JT, Shah DC, Garrigue S, Hocini M, Yamane T, et al. Left ventricular diastolic dysfunction in patients with so–called lone atrial fibrillation. J Cardiovasc Electrophysiol 2000;11(6):623–5. [ Links ]
15. Porthan KM, Melin JH, Kupila JT, Venho KK, Partinen MM. Prevalence of sleep apnea syndrome in lone atrial fibrillation: a case–control study. Chest 2004;125(3):879–85. [ Links ]
16. Sharifov OF, Fedorov VV, Beloshapko GG, Glukhov AV, Yushmanova AV, Rosenshtraukh LV. Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J Am Coll Cardiol 2004;43:483–90. [ Links ]
17. Mansour M, Mandapati R, Berenfeld O, Chen J, Samie FH, Jalife J. Left–to–right gradient of atrial frequencies during acute atrial fibrillation in the isolated sheep heart. Circulation 2001;103:2631–6. [ Links ]
18. Jalife J, Berenfeld O. Molecular mechanisms and global dynamics of fibrillation: an integrative approach to the underlying basis of vortex–like reentry. J Theor Biol 2004;230:475–87. [ Links ]
19. Cárdenas M, González Hermosillo A. Simposio fibrilación auricular: introducción. Arch Cardiol Mex 2004;74(Supl. 2):S287–S288. [ Links ]
20. Cárdenas M, Urina M, Sánchez A, Gaussí C. Fibrilación auricular y Wolf–Parkinson White. Arch Inst Cardiol Mex 1967;37:38–46. [ Links ]
21. Hsieh MH, Tai CT, Chiang CE, Tsai CF, Chen YJ, Chan P, et al. Double atrial potentials recorded in the coronary sinus in patients with Wolff–Parkinson–White syndrome: a possible mechanism of induced atrial fibrillation. J Interv Card Electrophysiol 2004; 11:97–103. [ Links ]
22. Goya M, Ouyang F, Ernst S, Volkmer M, Antz M, Kuck KH. Electroanatomic mapping and ablation of breakthroughs from the right atrium to the superior vena cava in patients with atrial fibrillation. Circulation 2002;106:1317–20. [ Links ]
23. Nanthakumar K, Lau YR, Plumb VJ, Epstein AE, Kay GN. Electrophysiological findings in adolescents with atrial fibrillation who have structurally normal hearts. Circulation 2004;110(2):117–23. [ Links ]
24. Todd DM, Skanes AC, Guiraudon G, Guiraudon C, Krahn AD, Yee R, Klein GJ. Role of the posterior left atrium and pulmonary veins in human lone atrial fibrillation: electrophysiological and pathological data from patients undergoing atrial fibrillation surgery. Circulation 2003;108(25):3108–14. [ Links ]
25. Haïssaguerre M, Jaïs P, Shah D, Takahashi A, Hocini M, Quiniou G, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 1998;339:659–66. [ Links ]
26. Iturralde P, Medeiros A, Guevara M, Kershenovich S, Varela S, Colín L. Fibrilación auricular focal tratada mediante radiofrecuencia. Arch Inst Cardiol Méx 2000;70:167–72. [ Links ]
27. Kalifa J, Jalife J, Zaitsev AV, Bagwe S, Warren M, Moreno J, et al. Intra–atrial pressure increases rate and organization of waves emanating from the superior pulmonary veins during atrial fibrillation. Circulation 2003;108:668–71. [ Links ]
28. Jais P, Hocini M, Macle L, Choi KJ, Deisenhofer I, Weerasooriya R, et al. Distinctive electrophysiological properties of pulmonary veins in patients with atrial fibrillation. Circulation 2002;106:2479–85. [ Links ]
29. Takahashi Y, Iesaka Y, Takahashi A, Goya M, Kobayashi K, Fujiwara H, et al. Reentrant tachycardia in pulmonary veins of patients with paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol 2003; 14:927–32. [ Links ]
30. Adragao P, Santos KR, Aguiar C, Neves JP, Abecassis M, Cavaco D, et al. Atrial fibrillation and effective refractory period of the pulmonary vein ostia. Rev Port Cardiol 2002;21 (10): 1125–34. [ Links ]
31. Perez–Lugones A, McMahon JT, Ratliff NB, Saliba WI, Schweikert RA, Marrouche NF, et al. Evidence of specialized conduction cells in human pulmonary veins of patients with atrial fibrillation. J Cardiovasc Electrophysiol 2003;14:803–9. [ Links ]
32. Tamargo J, Depón E. Nuevas terapéuticas diarias en el tratamiento de la fibrilación auricular. En: García E, Elizalde A, Onetti C, editores: Bases electrofisiológicas de las arritmias cardiacas.. Colima, Ed. Universidad de Colima 2009:54–68. [ Links ]
33. Weigl M, Gschwantler M, Gatterer E, Finsterer J, Stollberger C. Reflux esophagitis in the pathogenesis of paroxysmal atrial fibrillation: results of a pilot study. South Med J 2003; 96(11 ):1128–32. [ Links ]
34. Bettoni M, Zimmermann M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation 2002;105(23):2753–9. [ Links ]
35. Smeets JL, Allessie MA, Lammers WJ, Bonke FI, Hollen J. The wavelength of the cardiac impulse and reentrant arrhythmias in isolated rabbit atrium. The role of heart rate, autonomic transmitters, temperature, and potassium. Circ Res 1986;58:96–108. [ Links ]
36. Pappone C, Santinelli V, Manguso F, Vicedomini G, Gugliotta F, Augello G, et al. Pulmonary vein denervation enhances longterm benefit after circumferential ablation for paroxysmal atrial fibrillation. Circulation 2004;109:327–34. [ Links ]
37. Jaïs P, Hocini M, Sacher F, Clémenty J, Haïssaguerre M. The place of ablation in the treatment of atrial fibrillation: where are we and where are we going?. Arch Mal Coeur Vaiss 2004;97:1071–7. [ Links ]
38. Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 1997;96(4):1180–4. [ Links ]
39. Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, et al. C–reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation 2001;104(24):2886–91. [ Links ]
40. Sacher F, Corcuff JB, Schraub P, Le Bouffos V, Georges A, Jones SO, et al. Chronic atrial fibrillation ablation impact on endocrine and mechanical cardiac functions. Eur Heart J 2008;29:1290–5. [ Links ]
41. Nakamura Y, Nakamura K, Fukushima–Kusano K, Ohta K, Matsubara H, Hamuro T, et al. Tissue factor expression in atrial endothelia associated with nonvalvular atrial fibrillation: possible involvement in intracardiac thrombogenesis. Thromb Res 2003;111(3):137–42. [ Links ]
42. Schotten U, Duytschaever M, Ausma J, Eijsbouts S, Neuberger HR, Allessie M. Electrical and contractile remodeling during the first days of atrial fibrillation go hand in hand. Circulation 2003;107:1433–9. [ Links ]

