










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.79 no.2 Ciudad de México abr./jun. 2009
Artículo de investigación: aterosclerosis
Marcadores de estrés oxidativo en aterogénesis inducida por hiperfibrinogenemia
Oxidative stress markers in atherogenesis induced by hyperfibrinogenemia
María C Báez,1,2 Mariana D Táran,1,3 Vilma Campana,1,2 Juan C Simes,1 Patricia Pons,4 José Atilio–Palma,1 y Mónica Moya1,2*
1 Cátedra de Física Biomédica, Universidad Nacional Córdoba, Argentina.
2 Cátedra de Física Biomédica, Universidad Nacional La Rioja, Argentina.
3 Becaria Fundación Florencio Fiorini.
4 Centro Microscopia Electrónica, Facultad de Ciencias Médicas, Córdoba, Argentina.
* Autor para correspondencia:
Dra. Mónica Moya.
Cátedra de Física Biomédica. Santa Rosa 1085.
Córdoba. Código Postal 5000.
Facultad de Ciencias Médicas.
Universidad Nacional de Córdoba. Argentina.
Teléfono y Fax: 0351–4332020.
Correo electrónico: monicamoya@hotmail.com
Recibido el 20 de diciembre de 2007.
Aceptado el 3 de noviembre de 2008.
Resumen
Introducción: Estudiar el factor de necrosis tumoral alfa (TNF–α), el óxido nítrico (ON) y la citrulina en ratas con lesiones aterogénicas inducida por hiperfibrinogenemia, y su repercusión morfológica mitocondrial en el músculo liso aórtico.
Material y métodos: Inducción de hiperfibrinogenemia por inyección de adrenalina (0,1 mg/rata/día), estudiando: A) control, B) injuriados múltiples por 30 días y, C) injuriados múltiples por 60 días. Citrulina (mM) y ON (μM): espectrofotometría, TNF–α (pg/dL): Elisa y mitocondrias: microscopía electrónica. Los resultados se analizaron mediante: ANOVA, Coeficiente r y X2, estableciendo p < 0.05 para todos los casos.
Resultados: Se observó un incremento de TNF–α estadísticamente significativo tanto en (B) (50.05 ± 2.29) como en (C) (74.99 ± 2.82) respecto del control (A) (33.01 ± 1.49) (p < 0.001 en ambos grupos). Citrulina se comportó de manera similar, ya que aumentó significativamente en (B) (5.56 ± 0.20) y en (C) (6.84 ±0.13) comparado con (A) (4.41 ± 0.23) (p < 0.001 en ambas situaciones). La biodisponibilidad de ON disminuyó significativamente en (B) (8.97 ± 0.70) y en (C) (5.32 ± 0.68) al compararlo con el grupo (A) (21.65 ± 1.74) (p < 0.001 en ambos grupos).
Conclusiones: La hiperfibrinogenemia activaría la vía fisiopatológica del ON, produciría radicales libres y lesiones aterogénicas a nivel morfológico mitocondrial en el músculo liso de la aorta.
Palabras clave: Aterogénesis; Hiperfibrinogenemia; Estrés oxidativo; Mitocondria; Argentina.
Abstract
Introduction: We studied plasmatic TNF–α, nitric oxide (NO) and citrulline behaviors and probable morphological mitochondrial alterations in aortic smooth muscle cells, in rats with atherogenesis induced by hyperfibrinogenemia in: A) control, B) multiple injured for 30 days and C) multiple injured for 60 days.
Material and methods: Hyperfibrinogenemia induction: adrenaline injection (0,1mg/rat/day). TNF–α (pg/dL) was determined by Elisa and NO (uM) and citrulline (mM) by spectrophotometry. Morphological mitochondrial alterations were studied by electronic microscopy. Variables were analized: ANOVA, r coefficient and X2 test.
Results: We observed a significant increment of TNF–α in multiple injured for 30 days (B) (50.05±2.29) as well as in multiple injured for 60 days (C) (74.99±2.82) related to control (A) (33.01±1.49) (p <0.001 in both groups). Citrulline presented a significant increased in (B) (5.56±0.20) and (C) (6.84±0.13) when compared to (A) (4.41±0.23) (p <0.001 in both situations). Mean while NO biodisponibility diminished significantly in (B) (8.97±0.70) and in (C) (5.32±0.68) when compared to (A) (21.65±1.74) (p <0.001 in both situations).
Conclusions: Hyperfibrinogenemia could modify the NO physiopathological pathway and produced morphological mitochondrial alterations in aortic smooth muscle cells, probably producing ischemic lesions in the vascular wall and altering the vasodilatation response.
Key words: Atherogenesis; Hyperfibrinogenemia; Oxidative stress; Mitochondria; Argentine.
Introducción
Los factores de riesgo clásicos relacionados con la hemostasis, la inflamación y sistémicos han sido implicados en la iniciación y progresión de la disfunción endotelial presente en la aterogénesis.1 Sin embargo, existen factores emergentes que constituyen un factor de riesgo epidemiológicamente significativo en la aterogénesis, entre ellos la hiperfibrinogenemia, considerada como uno de los indicadores inflamatorios asociado a dicho proceso.23 Sin embargo, es aún desconocido cuál sería el mecanismo que utiliza el fibrinogeno para producir estas lesiones. En trabajos previos de nuestro laboratorio4 hemos demostrado que la hiperfibrinogenemia inducida por métodos experimentales genera en la pared de la aorta torácica de ratas lesiones tipo I, II y III aterogénicas, según la clasificación de Stary.5 Estos cambios reflejarían tanto la actividad inflamatoria de la progresión arterioesclerótica como la disfunción endotelial presente, permitiendo postular que el fibrinogeno, actuaría como un factor de riesgo con potencial aterogénico independiente desde los estadios iniciales de la enfermedad.6,7 Se ha postulado que el incremento de la síntesis de fibrinogeno estaría mediado por citocinas, entre ellas el factor de necrosis tumoral alfa (TNF–α), el cual promovería la activación endotelial,8 situación previa a la disfunción endotelial, condición sine qua non para la aterogénesis. Es probable que en este proceso el endotelio activado sintetice intermediarios reactivos del oxígeno: óxido nítrico (ON) y su co–producto: L–citrulina. Ambas moléculas serían sintetizadas en el interior de la célula endotelial a partir de una molécula de L–arginina, generándose así una reacción equimolar de L–citrulina y ON, catalizada por la enzima sintetasa del ON (ONS).9 También en la matriz mitocondrial se forma L–citrulina a partir de ornitina (Orn) y carbamoilfosfato, reacción catalizada por la ornitina transcarbamoilasa, teniendo la membrana interna mitocondrial un intercambiador de citrulina/ornitina, esta L–citrulina sintetizada es luego transportada al citosol. Cuando existen alteraciones del estrés oxidativo por estímulos proaterogénicos, como podría ser la hiperfibrinogenemia, estas moléculas de ON y L–citrulina podrían modificar su síntesis, y seguir una vía fisiopatológica, pudiendo transformarse en peroxinitritos o radicales de OH por la reacción con el anión superóxido (O2), produciendo así alteraciones en las mitocondrias del músculo liso vascular.10
El objetivo del presente trabajo es estudiar el mecanismo por el cual la hiperfibrinogenemia produce las alteraciones anatomopatológicas aterogénicas en la pared vascular aórtica, utilizando como indicadores del estrés oxidativo: a) la determinación de óxido nítrico y L–citrulina, y b) la probable alteración de la morfología mitocondrial en las células musculares lisas de la aorta torácica.
Material y métodos
Se utilizaron ratas machos adultos Cepa Suquía, de peso 280 ± 20 g, endocriadas en el Instituto de Fisiología de la Facultad de Ciencias Médicas de la Universidad Nacional de Córdoba, Argentina, durante el año 2006. La investigación fue realizada de acuerdo con la guía de cuidados y usos de animales de laboratorio publicado por el Instituto Nacional de Salud de Estados Unidos de América, publicación NIH (n° 85–23, revisado 1996). Los procedimientos utilizados en los animales han sido aprobados por la Comisión de Bioética de la Facultad de Ciencias Médicas, de la Universidad Nacional de Córdoba, Argentina.
Se utilizaron 36 animales divididos en los siguientes lotes de 12 animales cada uno:
A) Control (no injuriadas)
B) Injurias Múltiples por 30 días (IM × 30 ds)
C) Injurias Múltiples por 60 días (IM × 60 ds)
No se registraron muertes ni se excluyeron animales en ninguno de los lotes estudiados. Teniendo en cuenta trabajos previos de nuestro laboratorio,11 la inducción proinflamatoria se realizó por inyección subcutánea diaria de adrenalina (0.1 mg/rata) por períodos de 30 y 60 días posteriores a la 1ª inyección. La sangre se obtuvo por decapitación de los animales, a las 72 horas de la última injuria, coincidiendo con los 30 y 60 días. La sangre se recogió en cápsulas de Petri con una mezcla de anticoagulante constituido por oxalato de amonio y de potasio en una proporción de 2:1 e inmediatamente centrifugada a 3 000 rpm durante 15 minutos para obtener el plasma. El material para microscopia electrónica (ME) se obtuvo por cortes in toto en forma de anillos de 2 mm de aorta torácica en todos los lotes estudiados. El tejido fue fijado en Karnovsky12y las secciones ultrafinas, observadas y fotografiadas en un microscopio electrónico Leo 906E. Para valorar las características morfológicas de las mitocondrias se realizó una clasificación de 3 grados de acuerdo con el porcentaje de modificaciones mitocondriales que se observaron:
Grado 1: Apariencia normal con un tamaño de hasta 1.7 μm de tamaño.
Grado 2: Tamaño normal con crestas visibles pero escasas. Grado 3: Su tamaño es al menos el doble del normal. Mitocondrias muy dilatadas con alta desorganización de membranas interna y externa en algunas zonas, sin crestas visibles. Matriz clara con restos electrodensos próximos a membrana externa y presencia de vacuolas.
El TNF–α se analizó por técnica de ELISA ("Enzyme–Linked ImmunoSorbent Assay"– Ensayo por inmunoabsorción ligado a enzimas), utilizando un juego de reactivo (Betnoff D),13 expresando los resultados en pg/dL. El ON se dosificó por reacción de Griess por espectrofotometría, expresando los resultados en μM.14 La L–citrulina se determinó por método colorimétrico y se expresaron los resultados en mM.15
Para descartar el efecto del estrés y su repercusión en los diversos órganos y sistemas, se determinó adrenalina plasmática en todos los grupos estudiados. En el lote control se realizó inyección subcutánea diaria de solución fisiológica (0.1 mg/rata) para sistematizar los mecanismos de intervención a todos los lotes, y la medición se realizó por cromatografía líquida con detección electroquímica16 (HPLC–DE, por sus siglas en inglés) y los resultados se expresaron en pg/mL.
Los resultados de las variables continuas se analizaron con un modelo de ANOVA. La asociación entre las varia0bles cuantitativas fue estudiada a través del coeficiente "r" de Pearson y el coeficiente de determinación (r2). Para la cuantificación mitocondrial se utilizó el programa Axio–visión 3.0, y para el análisis de la cuantificación se utilizó χ2. Se estableció un nivel de significancia de p < < 0.05 para todos los casos.
Resultados
Los resultados de los niveles plasmáticos del TNF–α se muestran en la Figura 1.
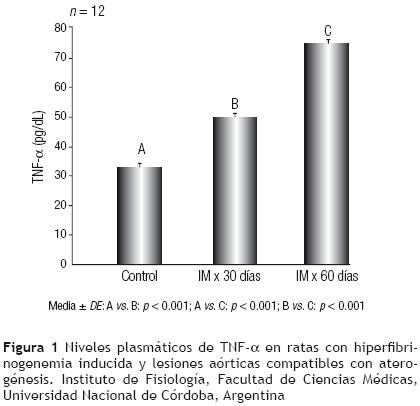
Las concentraciones del TNF–α demostraron un incremento significativo en los lotes IM × 30 días y en IM × 60 días al ser comparados con el lote control (p < 0.001 para ambos grupos).
Los resultados de los niveles plasmáticos de ON se muestran en la Figura 2.

Se observó una disminución estadísticamente significativa en las concentraciones de ON en el grupo de IM por 30 días (B) respecto del control (A) (p < 0.001). Similar comportamiento se demostró cuando se comparó el grupo de IM × 60 días (C) respecto del control (A) (p < 0.001). Se advirtió que a medida que la hiperfibrinogenemia persistió en el tiempo, el nivel de ON disminuía significativamente, resultados que se evidencian cuando se compara el grupo (C) con el (B) (p < 0.001).
Los resultados de las concentraciones plasmáticas de la L–citrulina se muestran en la Figura 3.

Otras de las variables estudiadas en relación con el posible endotelio activado por hiperfibrinogenemia es la L–citrulina, co–producto de la reacción del ON, por lo cual se esperaría que los niveles de L–citrulina se comportaran como el ON. Sin embargo, se observó que la concentración de L–citrulina aumentó significativamente en los lotes IM × 30 días (B) e IM × 60 días (C) en comparación con el control (A) (< 0.001); además, se observó un incremento significativo entre el grupo (C) respecto del (B) (p < 0.001).
La correlación entre la hiperfibrinogenemia y los marcadores de estrés oxidativo se muestran en la Figura 4.

Cuando se analizaron los coeficientes de correlación entre las distintas variables se observó que existe una correlación perfecta (r y r2 =1) entre los valores de fibrinógeno y TNF–α; una correlación inversa entre FP y ON (r = –0.78 y r2 = 0.61); y una correlación directa entre FP y L–citrulina (r = 0.75 y r2 = 0.56). Estos resultados sugieren que la hiperfibrinogenemia se correlaciona con los marcadores de estrés oxidativo.
Los resultados de las mediciones mitocondriales por ME en el músculo liso de la aorta se muestran en la Tabla 1.

Cuando se analizaron los resultados morfológicos mitocondriales en el músculo liso de la aorta torácica por ME, a medida que la hiperfibrinogenemia incrementa por inducción de injurias múltiples (IM) y el ON disminuye, se observó que el número total de mitocondrias disminuyó significativamente comparando los grupos (B) (Figuras 5B y 5C), (Figura 5C) con el grupo control (Figura 5A) (p < 0.01).
Similar comportamiento presentó el número total y medio de las mitocondrias que disminuyeron significativamente en los grupos (B) y (C), asociado con hiperfibrinogenemia y ON disminuido en comparación con el control (A) (p < 0.01).
Si bien los diámetros mitocondriales de todos los lotes analizados no se modificaron, se observó en los lotes (B) y (C) una notable dilatación del espacio intermembranoso y también de las crestas mitocondriales que se corresponderían a lesiones compatibles con tumefacción mitocondrial. Por otra parte, tanto el área total como el área media de las mitocondrias de los grupos sometidos a IM por 30 días (B) y por 60 días (C), disminuyeron significativamente en relación con el grupo control (A) (p < 0.01), dado que las mitocondrias tienden en los grupos (B) (Figura 5B) y (C) (Figura 5C) a ubicarse en formas agrupadas en proximidades al núcleo, presentando aclaramiento de la matriz con vacuolas y crestas desorganizadas e incluso algunas sin crestas (Figuras 5B y 5C). Además, cuando se compararon las alteraciones morfológicas entre los grupos IM por 30 días (B) (Figura 5B), y por 60 días (C) (Figura 5C), se observó en este último grupo la presencia de acúmulos de gránulos electrodensos localizados en la matriz mitocondrial, que se corresponderían a depósitos citosólicos de Ca+2 que habitualmente se encuentra secuestrado en las mitocondrias y en el retículo endoplásmico. Se observaron algunas alteraciones en el grupo control, que son atribuidas a que la mitocondria es un organelo lábil, y que aun con la técnica de extracción más sensible presenta un porcentaje de cambios morfológicos grado I (7.77%), semejante al obtenido en nuestro modelo experimental.
Se destacó que al comparar el grado 3 de alteración se observa que en el grupo IM × 60 días (C) (94.21%) las lesiones de este tipo son significativamente mayores respecto a los grupos IM × 30 días (B) (73.41%) (p < 0.01). Todos los cambios mitocondriales descritos se corresponden al tipo de configuración ortodoxa.
Los resultados de adrenalina plasmática se presentan en la Figura 6.

En los grupos IM por 60 días (C) se observó una disminución significativa de los niveles de adrenalina con respecto al grupo (B) y al grupo control (A) (p < 0.001). Similar comportamiento se observó al realizar la comparación del grupo IM por 30 días (B) respecto al grupo (C) (p < 0.001).
Discusión
Los niveles de TNF–α aumentados significativamente en los grupos con hiperfibrinogenemia inducida indican la existencia de activación endotelial. El TNF–α se comportaría como un marcador bioquímico del grado de disfunción endotelial en relación con la injuria vascular.17
La disminución de ON en los grupos con hiperfibrinogenemia inducida permitiría inferir que actuaría como factor proaterogénico, generando estrés oxidativo, con probable pérdida de la capacidad de las células endoteliales para regular sus funciones vitales en la producción de sustancias vasoactivas.9 Probablemente la disminución en la síntesis de ON se acompañe de aumento de radicales libres, como peroxinitrito (OONO–) y anión superóxido (O–), cuyas concentraciones se encuentran elevadas generando una menor biodisponibilidad de ON, lo cual provoca alteraciones en la respuesta vasodilatadora, función que se lleva a cabo a través del ON,18 generando alteraciones de flujo vascular e isquemia.
Por lo tanto, la disminución de ON debería tomarse en cuenta como marcador significativo de estrés oxida tivo en las células endoteliales durante la iniciación de la aterogénesis,19 y actuaría en pacientes como predictor del riesgo para futuros eventos cardiovasculares de tipo isquémico.
Respecto a los niveles plasmáticos de L–citrulina en situación de hiperfibrinogenemia, a pesar de que su producción es equimolar respecto a la producción de ON, la misma aumentó significativamente en el plasma. Esto podría deberse a que la L–citrulina seguiría una ruta diferente a la del ON,20 conservando el rol antiproliferativo y vasodilatador. Posiblemente el incremento plasmático de L–citrulina sea un mecanismo fisiológico mediante el cual se intenta compensar la baja biodisponibilidad del ON en el proceso aterogénico, mecanismo que merece mayor investigación.21
Las modificaciones del estrés oxidativo por hiperfibrinogenemia inducida4,8 impactarían a nivel mitocondrial con cambios morfológicos de tipo ortodoxo como observamos en nuestros resultados. Los cambios en los grupos (B) y (C) podrían ser debidos a la entrada del ión Ca+2 al citosol, lo cual genera depósitos de sales insolubles y tumefacción turbia;22 bajo estas condiciones los gránulos de la matriz desaparecen, conservando una apariencia clara. Asociado a las alteraciones descriptas se induce a un incremento del área mitocondrial debido al aumento de agua en la matriz de dicho organelo como consecuencia de la alteración en la integridad de la membrana celular, ello afecta su permeabilidad así como la homeostasis iónica plasmática, tanto de la célula como de la mitocondria. Por otra parte, se observó que seguida a la tumefacción turbia descripta se produjo una desaparición de las crestas mitocondriales denominada cristólisis; tal lesión es reversible en su fase inicial.23
Los procesos mitocondriales mencionados serían compatibles con alteraciones funcionales, ya que en la membrana interna de la mitocondria se localiza la cadena de transporte de electrones donde se generan los radicales libres del oxígeno, los cuales en concentraciones elevadas provocarían el deterioro del organelo por estrés oxidativo.24–27 La configuración morfológica de tipo ortodoxa observada en los grupos injuriados corresponden a una configuración que se asocia a un bajo nivel de fosforilación oxidativa, situación que se relaciona con la lesión isquémica, donde se interrumpe la incorporación de oxígeno en la cadena de transporte de electrones, resultando así un efecto adverso, por la producción masiva de especies reactivas de oxígeno (ROS, por sus siglas en inglés) para compensar la falta de oxígeno en la respiración aeróbica mitocondrial,23 tal como se ha estudiado en otro trabajo.28 Como consecuencia, se puede inferir que se iniciará la disfunción mitocondrial, induciendo de esta forma a que se genere una menor producción de ATP en dicho organelo, alterando la morfología y probablemente la actividad funcional, lo cual perpetuaría la lesión isquémica patognomónica de la aterogénesis.29
Por lo tanto, la baja disponibilidad de ON presente en los grupos injuriados, asociado a las alteraciones morfológicas mitocondriales en el músculo liso vascular, demostrarían la disfunción endotelial en la aterogénesis inducida por la hiperfibrinogenemia.
Los resultados de adrenalina plasmática demostrarían que la liberación de la misma por parte de la médula suprarrenal se encuentra inhibida, posiblemente a causa de un sistema tipo down–regulation, ya sea por una disminución del número de receptores disponibles o su inactivación por el mecanismo de inducción proinflamatorio utilizado.30 Además, los valores de adrenalina nos demuestran que tanto las lesiones observadas en la pared vascular de la aorta torácica, como las variaciones de los biomarcadores estudiados y las alteraciones morfológicas en las mitocondrias del músculo liso vascular aórtico son inducidas por la hiperfibrinogenemia y por su impacto en el estrés oxidativo a nivel de la aorta, y no por los efectos de la adrenalina.
Los datos obtenidos nos mostrarían que la hiperfibrinogenemia generaría estrés oxidativo a nivel endotelial repercutiendo sobre la mitocondria y alterando su morfología, y probablemente modificaría su actividad funcional, reflejando así el tipo de lesión isquémica cuya expresión fisiopatológica es la aterogénesis.
Financiado por subsidios de la Fundación Roemmers y Secretaría de Ciencia y Técnica de la Universidad de Córdoba y Universidad de La Rioja, Argentina.
Bibliografía
1. Tzouloki I, Murray GD, Price JF, Smith FB, et al. Hemostatic factors, inflammatory markers, and progressive peripheral atherosclerosis: The Edinburgh artery study. Am J Epidemiol 2006;163(4):334–41. [ Links ]
2. Drouet L, Baldit Sollier C. Is fibrinogen a predictor or a marker of the risk of cardiovascular events? Therapie 2005;60(2):125–36. [ Links ]
3. Izaguirre–Ávila R, Zaldívar–Alcántara H. El fibrinógeno como factor de riesgo cardiovascular. Arch Cardiol Mex 2003;73:7–10. [ Links ]
4. Moya M, Campana V, Gavotto A, et al. Hyperfibrinogenemia in rats treated with meloxicam. Jpn Heart J 2002;43(5):559–66. [ Links ]
5. Fuster V, Fayard ZA, Badimon S. Acute coronary syndrome: biology. Lancet 1999;353:5–9. [ Links ]
6. Kamath S, Lip GYH. Fibrinogen: biochemistry, epidemiology and determinants. QJMed 2003;96:711–29. [ Links ]
7. Jerjes–Sánchez Díaz C, Comparan–Núñez A, Ibarra–Flores M, Decanini–Arcaute H, et al. Marcadores hemostáticos y de inflamación en síndromes coronarios agudos y su asociación con eventos cardiovasculares adversos. Arch Cardiol Mex 2006; 76(4):366–75. [ Links ]
8. Moya M, Campana V, Gavotto A, Spitale L, et al. Simvastatin: pharmacological response in experimental hyperfibrinogenemias. Acta Cardiol 2005;60(2):159–64. [ Links ]
9. Kleinbongard P, Dejam A, Lauer T, JaxT, et al. Plasma nitrite concentrations reflect the degree of endothelial dysfunction in humans. Free Radic Biol Med 2006;40(2):295–302. [ Links ]
10. Yeong–Renn C, Chwen–Lih C, Alexander Y et al. Direct and indirect roles of cytochrome b in the mediation of superoxide generation and ON catabolism by mitochondrial succinate–cytochrome c reductase. J Biol Chem 2006;281 (19):13159–68. [ Links ]
11. Palma JA, Enders J, de Oliva PP. Effects of epinephrine on plasma fibrinogen levels in rats submitted to tissue injury. Experientia 1981;37(7):780–2. [ Links ]
12. Karnovsky MJ, Graham RC. A formaldehide–glutaraldehide fixative of high osmolarity by MET. J Cell Biol 1965;27:137–8. [ Links ]
13. Betnoff D, Benoliel AM, Peiretii F, Lopez S, et al. Effect of atorvastatin on adhesive phenotype of human endotelial cells activated by tumor necrosis factor alpha. J Cardiovasc Pharmacol 2003;41(2):316–24. [ Links ]
14. Schulz K, Kerber S, Kelm M. Reevaluation of the Griess method for determining NO/NO2– in aqueous and protein–containing samples. Nitric Oxide. 1999 Jun;3(3):225–34. [ Links ]
15. Boyde TRC, Rahmatullah M. Optimization of conditions for the colorimetric determination citrulline, using diacetyl monoxime. Anal Biochem 1980;107(2):424–31. [ Links ]
16. Garcia–Rudoz C, Armando I, Levin G, Escobar ME, et al. Peripheral catecholamine alterations in adolescents with polycystic ovary síndrome. Clin Endocrinol 1998;49(2):221–8. [ Links ]
17. Libby P. Vascular biology of atherosclerosis: overview and state of the art. Am J Cardiol 2003;91:3A–6A. [ Links ]
18. Yang Z, Ming X. Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin Med Res 2006;4(1 ):53–65. [ Links ]
19. Guzik TJ, West NEJ, Pillai R, Taggart DP, et al. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension 2002; 39:1088–94. [ Links ]
20. Lind L. Circulating markers of inflammation and atherosclerosis. Atherosclerosis 2003;169:203–14. [ Links ]
21. Hayashi T, Packiasamy AR, Matsui–Hirai H, Miyazaki A, et al. L–citrulline and l–arginine supplementation retards the progression of high–cholesterol–diet–induced atherosclerosis in rabbits. Pharmacology 2005;102(38):13681–6. [ Links ]
22. Giulivi C. Characterization and function of mitochondrial nitricoxide synthase. Free Radic Biol Med 2003;34(4):397–408. [ Links ]
23. Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci 2005;26(4):190–5. [ Links ]
24. Boveris DL, Boveris A. Oxygen delivery to the tissues and mitochondrial respiration. Front Biosci 2007; 12:1014–23. [ Links ]
25. Chuaqui B, Duarte I, González S, Rosenberg H. Manual de Patología General. 2a edición. Santiago de Chile: Ediciones Universidad Católica de Chile; 1999 [consultado 2009 junio 21]. Disponible en: http://escuela.med.puc.cl/publ/PatologiaGeneral/ManualPatologiaIndice.html) [ Links ]
26. Martínez J. Mecanismos del daño celular en la insuficiencia renal aguda. Iatreia 1989;2(1):64–7. [ Links ]
27. Proshchaev KI, IInitskii AN, Kvetnoi IM, Kniazkin IV, et al. Changes of the endothelium in cardiovascular pathology in the elderly. Part I. Signal molecules and endothelial functions. Klin Med (Mosk) 2007;85(11):9–13. [ Links ]
28. Tarán M, Baez M, Campana V, Simes J, et al. Valoración de la respuesta farmacológica de diclofenaco y gemfibrozil en un modelo experimental aterogénico por hiperfibrinogenemia inducida en ratas. Medicina 2007;67(III):219. [ Links ]
29. Giordano F. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 2005;115(3):500–8. [ Links ]
30. Osadchii OE, Norton GR, McKechnie R, Deftereos D, et al. Cardiac dilatation and pump dysfunction without intrinsic myocardial systolic failure following chronic beta adrenoreceptor activation. Am J Physiol Heart Circ Physiol 2007;292(4):H1898–905. [ Links ]

