










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.77 no.3 Ciudad de México jul./sep. 2007
Editorial
Entendiendo la hipertrofia ventricular izquierda
Understanding left ventricular hypertrophy
José Fernando Guadalajara Boo*
* Editor en Jefe Archivos de Cardiología de México.
Correspondencia:
Dr. José Fernando Guadalajara Boo.
Instituto Nacional de Cardiología Ignacio Chávez
(INCICH, Juan Badiano Núm. 1. Col. Sección XVI, Tlalpan
14080, México, D.F.).
Tel. 56 55 29 24. Fax. 55 73 09 94.
E-mail: guadalajara@cardiologia.org.mx
Recibido: 20 de junio de 2007
Aceptado: 22 de junio de 2007
Palabras clave: Hipertrofia adecuada. Hipertrofia inadecuada. Hipertrofia patológica.
Key words: Adequate hypertrophy. Inadequate hypertrophy. Pathologic hypertrophy.
Se ha demostrado en forma fehaciente que la hipertrofia ventricular izquierda, representa un importante factor de riesgo de mortalidad en la población general,1-5 en hipertensión arterial6,7 en la estenosis aórtica grave;8 en la insuficiencia cardíaca,9 y en aquellos pacientes afectados por cardiopatía isquémica,10,11 lo cual contrasta, con la hipertrofia fisiológica que determina el crecimiento del corazón durante el desarrollo del ser humano, la hipertrofia que acompaña el embarazo y al corazón del atleta, la cual no tiene consecuencias patológicas.13-15 Es por ello que Escudero y Pinilla llaman la atención sobre este hecho en el artículo <<Paradigma y paradojas de la hipertrofia ventricular izquierda: desde el laboratorio de investigación a la consulta clínica>>, publicado en este número16 y el cual revela las reflexiones de este grupo de investigadores cuya línea de estudio ha sido la hipertrofia ventricular izquierda, que se demuestra por el extenso número de publicaciones que tiene al respecto y por lo que esta publicación merece un comentario editorial. En efecto, ¿cómo entender y explicar la paradoja de la hipertrofia ventricular izquierda en la que, por un lado, se muestra como un proceso adaptativo que ayuda el funcionamiento ventricular y, por otro, es causa de mortalidad? y ¿cómo entender la génesis de hipertrofia fisiológica y la posibilidad de transformar la patología en fisiología?, reflexiones con las que los autores culminan su conclusión.
Cuando se analiza la función que cumple la hipertrofia en la fisiología del corazón adulto, se puede concluir que es un mecanismo que aparece como consecuencia de los procesos de remodelación ventricular17 para normalizar el estrés sistólico en las sobrecargas de presión (hipertensión arterial y estenosis aórtica)18,19 (Fig. 1) y el estrés diastólico en las sobrecargas de volumen (insuficiencia aórtica)20 (Tabla I).


En las primeras, el gasto cardíaco se mantiene, a pesar de que el corazón se vacía ante una presión significativamente mayor, y como el estrés sistólico (postcarga), es también uno de los más importantes determinantes del MVO221 al ser normalizado por el mayor engrosamiento miocárdico durante la sístole, el aumento de la función se lleva a cabo sin mayor costo metabólico. Asimismo, en las sobrecargas de volumen, la hipertrofia permite normalizar el estrés diastólico20,22 (Tabla I) y, con ello, el corazón puede desplazar una mayor cantidad de volumen diastólico y evitar que la sobrecarga lleve el corazón hacia la insuficiencia cardíaca.20,22
Vale la pena enfatizar que tanto en las sobrecargas de presión como en los de volumen, el aumento del crecimiento miocítico se acompaña de un crecimiento intersticial concordante (Fig. 2), lo cual ofrece un soporte estructural a la hipertrofia. Este crecimiento congruente entre la masa miocítica y la colágena intersticial, permite al corazón aumentar la función para enfrentar la sobrecarga, con la característica fundamental de no activar el sistema neuroendocrino. De esta manera, la hipertrofia miocárdica se puede considerar como un eficiente mecanismo de compensación para el corazón,23 el cual le permite por un lado mantener una función normal sin aumentar el consumo de oxígeno miocárdico (MVO2),21 a pesar del aumento de la fuerza contráctil que provee la hipertrofia compensadora (Fig. 3) y todo ello se lleva a cabo sin estimular mecanismos neuro-humorales. En estas condiciones, a la hipertrofia se le denomina <<hipertrofia adecuada>>.24


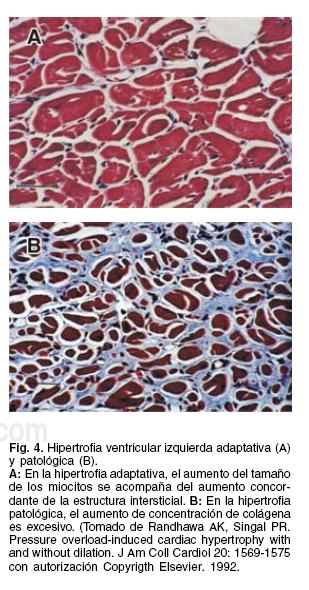
En clínica, es posible reconocer la hipertrofia adecuada en los casos de insuficiencia aórtica, cuando es capaz de normalizar el estrés diastólico (Tabla I), o sea, cuando la relación grosor/ radio (h/r) en diastole se mantiene en cifras normales (0.47 ± 0.05),25,26 en presencia de dilatación ventricular izquierda.22 En el caso de la estenosis aórtica y en la hipertensión arterial, la hipertrofia es adecuada cuando el corazón es capaz de normalizar el estrés sistólico, a pesar de existir una presión intraventricular izquierda muy elevada18,19 (Fig. 1) y ello sólo puede lograrse con un engrosamiento sistólico muy importante, que concomitantemente reduce en forma muy significativa el radio sistólico de la cavidad ventricular (Fig. 3), por un aumento muy considerable de la fuerza contráctil. Este hecho coincide con los hallazgos experimentales de Randhawa y col27, en cobayos con hipertrofia adaptativa demostrada histológicamente (Fig. 4), en los que se encuentra aumento del dp/dt.
La diferencia entre la hipertrofia compensadora de la insuficiencia aórtica,22 en relación con la estenosis aórtica e hipertensión arterial,18,19 es que, en la primera, la hipertrofia adecuada se manifiesta por una relación grosor/radio en diastole con valores normales,22,25 mientras que en las sobrecargas de presión el grado de hipertrofia, que se requiere para normalizar la postcarga, es muy superior a los valores normales (0.70 ± 0.16 para la hipertensión arterial y 0.97 ± 0.27 para estenosis aórtica), por lo que cae en la categoría de hipertrofia inapropiada.18,19,24
El mecanismo de la hipertrofia en estas condiciones, se puede considerar como el más importante para evitar que el corazón sobrecargado presente insuficiencia cardíaca. La hipertrofia miocárdica puede compensar la sobrecarga por mucho tiempo, a veces por años. Sin embargo, cuando la sobrecarga hemodinámica importante se perpetúa en el tiempo, la hipertrofia puede llegar a ser insuficiente para normalizar el estrés diastólico tal y como sucede en la insuficiencia aórtica (Tabla I), lo cual se manifiesta porque el radio de la cavidad excede en proporción al espesor de la pared y se reduce la relación grosor/radio (h/r) en diastole (hipertrofia inadecuada o hipertrofia excéntrica),22-24 aumenta el estrés diastólico y con ello el engrasamiento sistólico es insuficiente para normalizar el estrés sistólico (postcarga).20,22 El ejemplo más evidente de hipertrofia inadecuada es la miocardiopatía dilatada, y el papel que la hipertrofia ventricular juega en esta enfermedad es muy importante, ya que se ha demostrado que, entre menos hipertrofia exista (menor valor de la relación h/r en diastole), es mayor la mortalidad anual.26 Debido a la relación inversa que tiene la postcarga con la función ventricular28,29 al aumentar la primera, se reduce la fracción de expulsión paulatinamente hasta llegar a la insuficiencia cardíaca. En esta fase, se activa el sistema neuroendocrino y si ésta, a su vez, se prolonga en el tiempo, la acción sostenida de la angiotensina II y de la aldosterona favorece que en el intersticio miocárdico se aumente excesivamente la colágena27,30-33 (Fig. 4); y, en estas condiciones, la hipertrofia se transforma paulatinamente de proceso adaptativo a proceso pato-lógico23,27,30-32 ya que no es capaz de normalizar la precarga y participa en la progresión de la insuficiencia cardíaca. En lo referente a las sobrecargas de presión (estenosis aórtica e hipertensión arterial), la hipertrofia miocárdica, como se mencionó, también juega un papel preponderante en la compensación hemodinámica de la sobrecarga sistólica (Fig. 1).19 Pero, al igual que en la insuficiencia aórtica, cuando la sobrecarga se sostiene en el tiempo, el aumento excesivo de colágena intersticial,27 (Fig. 4) inicialmente reduce la distensibilidad ventricular y condiciona disfunción diastólica.32,33 Cuando el contenido de colágena supera el 200% de sus concentraciones normales, aparece insuficiencia cardíaca27,32 y es posible que a esta fase corresponda el hallazgo de Escudero y col. en el que demuestra menor capacidad contráctil de corazón de las ratas hipertensas.34 En efecto, cuando la hipertrofia es insuficiente para normalizar el estrés sistólico (postcarga), la fracción de expulsión se reduce paulatinamente hasta llegar a la insuficiencia cardíaca (Fig. 1).29 Cuando la insuficiencia cardíaca es debida a una postcarga excesiva sin daño miocárdico, tal y como puede suceder en la estenosis aórtica, el tratamiento quirúrgico (se reduce la postcarga excesiva) produce una mejoría espectacular de la insuficiencia cardíaca.35 Sin embargo, tanto en la hipertensión arterial como en la estenosis aórtica, cuando la hipertrofia se acompaña de un aumento importante de la concentración de colágena intersticial (Fig. 4), la hipertrofia no sólo imposibilita la contracción suficiente para normalizar el estrés sistólico, sino que produce múltiples trastornos en la circulación coronaria, aumenta la rigidez ventricular, condiciona trastornos de la conducción y del ritmo, y la suma de todos estos cambios estructurales y funcionales, transformará la hipertrofia en un proceso patológico27,30-34 (Fig. 2) que en el caso de la hipertensión arterial constituye la <<Cardiopatía-hipertensiva>>.30-32 En la estenosis aórtica, también se ha demostrado que el daño estructural del miocardio es causado por la activación crónica del sistema RAA;8,30,-32,36 así, en la estenosis aórtica con insuficiencia cardíaca, el déficit de contracción miocárdica puede ser debido a sobrecarga excesiva o a daño miocárdico intrínseco, en este último caso, no se alivia la insuficiencia cardíaca al reducir la postcarga con la cirugía valvular aórtica.35
Si bajo estos preceptos, analizamos los estudios que demuestran que la hipertrofia miocárdica es un marcador de mortalidad,1-9,11,12 podremos entender que se refieren a hipertrofia patológica30,33,36 (Fig. 4) y no es que necesariamente la hipertrofia miocárdica siempre represente una <<mala adaptación>> a la sobrecarga, ni que la hipertrofia sea un proceso necesariamente anormal, y siempre sea un marcador de enfermedad y muerte. Más aún, la falta de hipertrofia y de crecimiento de colágena intersticial congruente en la insuficiencia mitral crónica, son los responsable de que aparezca daño miocárdico imperceptible, porque esta valvulopatía no desarrolla hipertrofia tempranamente en su evolución, debido a que funciona con postcarga baja37 y ello da lugar a que la presencia de hipertrofia inadecuada (relación h/r en diastole 0.36 ± 0.05) coincida con fracción de expulsión normal.38,39 Este comportamiento es el responsable de la evolución postoperatoria significativamente menos buena en relación con la insuficiencia aórtica.40,41 Al igual que la insuficiencia mitral crónica, en la mayoría de los casos de infarto del miocardio anterior extenso transmural, la hipertrofia de la pared posterior, que intenta ser compensadora, no logra normalizar el estrés diastólico ni sistólico, lo cual da lugar a la remodelación ventricular patológica progresiva.42 Con respecto a la miocardiopatía hipertrófica, realmente es una enfermedad miocárdica primaria, no es un mecanismo adaptativo y característicamente es una hipertrofia patológica que no responde a la necesidad de normalizar el estrés diastólico o sistólico del corazón de estos enfermos,43 por lo cual se comporta como una hipertrofia inapropiada24 con las consecuencias a las que ésta da lugar y sin los beneficios que ofrece la hipertrofia en un corazón con sobrecarga hemodinámica.
Finalmente, la posibilidad de lograr regresión de la hipertrofia al reducir la carga hemodinámica excesiva,44 incluso con reducción de colágena intersticial, al bloquear los efectos de la angiotensina II con inhibidores de la Eca44-47 o de la aldosterona (espironolactona o eplerrenona), preservando intacta la función ventricular, se ha denominado <<cardiorreparación>>.45 Estos conceptos adquieren importancia clínica, ya que al lograrse la regresión de la hipertrofia adaptativa o patológica, es posible cambiar el pronóstico para la vida de estos enfermos.
En conclusión, la hipertrofia adaptativa es un eficiente mecanismo de compensación, que permite mantener vivo y en condiciones de normalidad la función cardíaca por largos periodos de tiempo, defiende al paciente de la insuficiencia cardíaca y de la muerte, puede haber regresión de la hipertrofia cuando la causa se corrige oportunamente. La falta de hipertrofia adaptativa es nociva en los pacientes con insuficiencia mitral,28,37,38,40 miocardiopatía dilatada26 e infarto transmural extenso,42 porque al no defender al corazón de la remodelación patológica, aparece insuficiencia cardiaca progresiva que es una alta proporción de estos pacientes evolucionan hacia la muerte. Por otro lado, la progresión hacia la hipertrofia patológica es temible, porque es un marcador muy importante para mortalidad cardiovascular.1-11 Las aportaciones de Escudero y Padilla,16 en conjunto con todas las de los otros investigadores que han ofrecido conocimientos para el entendimiento de la función y significado de la hipertrofia ventricular izquierda, son contribuciones que nos ayudan a tener una interpretación panorámica más adecuada para aplicarla en la clínica a la cabecera del enfermo.
Referencias
1. Levy D, Anderson KM, Savage D, Kannel WB, Christiansen JC, Castelli WP: Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors. The Framingham Heart Study. Ann Intern Med 1988; 108: 7-13. [ Links ]
2. Casale PN, Devereux RB, Milner M, Zullo G, Harshfield GA, Pickering TG, Laragh JH: Value of echocardio graphic measurement of left ventricular mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med 1986; 105: 173-178. [ Links ]
3. Levy D. Garrison RJ, Savage DD, Kannel WB, Castelli WP: Prognostic implications of echocardio graphically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322: 1561-1566. [ Links ]
4. Kannel WB, Dannenberg AL, Levy D: Population implications of electrocardiographic left ventricular hypertrophy. Am J Cardiol, 1987; 60: 851-931. [ Links ]
5. Savage DD, Garrison RJ, Kannel WB, Levy D, Anderson SJ, Stokes J, et al: The spectrum of left ventricular hypertrophy in a general population sample: The Framingham Study. Circulation 1987; 75: 126-133. [ Links ]
6. Koren MJ, Devereux RB, Cásale PN, Savage DD, Laragh JH: Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345-352. [ Links ]
7. Schillaci F, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F: Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000; 35: 580-586. [ Links ]
8. Lund O: Preoperative risk evaluation and stratification of long-term survival after valve replacement for aortic stenosis. Reasons for earlier operative intervention. Circulation 1990; 82: 124-34. [ Links ]
9. Quiñones MA, Greenberg BH, Kopelen HA, Koilpillai C, Limacher MC: Echocardiographic Predictors of Clinical Outcome in Patients With Left Ventricular Dysfunction Enrolled in the SOLVD Registry and Trials: Significance of Left Ventricular Hypertrophy. J Am Coll Cardiol 2000; 35: 1237-44. [ Links ]
10. Cooper RS, Simmons BE, Castaner A, Santhanam V, Ghali J, Mar M: Left ventricular hypertrophy is associatedwith worse survival independent of ventricular function and number of coronary arteries severely narrowed. Am J Cardiol 1990; 65: 441-445. [ Links ]
11. Ghali JK, Liao Y, Simmons B, Castaner A, Cao G, Cooper RS: The prognostic role of left ventricular hypertrophy in patients with or without coronary artery disease. Ann Intern Med 1992; 117: 831-836. [ Links ]
12. Ghali JK, Liao Y, Cooper RS: Influence of left ventricular geometric patterns on prognosis in patients with or without coronary artery disease. J Am Coll Cardiol 1998; 31: 1635-1640. [ Links ]
13. Brown AJ Jr: Morphologic factors in cardiac hypertrophy. In: Alpert NR, ed. Cardiac hypertrophy. New York: Academic Press. 1971: 11. [ Links ]
14. Colan SD, Sanders SP, Borow KM: Physiopathologic hypertrophy: Effects on left ventricular systolic mechanics in athletes. J Am Coll Cardiol 1987; 9: 776-83. [ Links ]
15. Bar-Shlomo BZ, DruckMN, Morch JE: Left ventricular function in trained and untrained healthy subjects. Circulation 1982; 65: 484-488. [ Links ]
16. Escudero EM, Pinilla OA: Paradigmas y paradojas de la hipertrofia ventricular izquierda: desde el laboratorio de investigación a la consulta clínica. Arch Cardiol Mex 2007: 77. [ Links ]
17. Guadalajara JF, Alexanderson E: Remodelación ventricular. Editorial. Arch Inst Cardiol Mex 1993; 63: 85-8. [ Links ]
18. Guadalajara JF, Galván MO, Camacho P, Espínola N, Cervantes J, Huerta D: Cambios estructurales y funcionales en el corazón del hipertenso. Estudio ecocardiográfico. Arch Inst Cardiol Mex 1995; 65: 31-38. [ Links ]
19. Guadalajara JF, Martínez SC, Huerta D: La relación grosor/radio del ventrículo izquierdo (h/ r) en pacientes con estenosis aórtica. Implicaciones pronosticas y terapéuticas. Arch Inst Cardiol Mex 1990; 60: 383-391. [ Links ]
20. Osbakken M, Bove A, Span JF: Left ventricular function in chronic aortic regurgitation with reference to systolic pressure/volume and stress relations. Am J Cardiol 1981; 47: 193-8. [ Links ]
21. Weber KT, Janicki JS: Myocardial oxygen consumption the role of wall force and shortening. Am J Physiol 1977; 233: H421-30. [ Links ]
22. Guadalajara JF, Gual J, Martínez SC, Monobe F, Alexanderson, Cervantes JL: La hipertrofia miocárdica en la insuficiencia aórtica como mecanismo de compensación. Implicaciones para la indicación quirúrgica. Arch Inst Cardiol Mex 1992; 62: 351-360. [ Links ]
23. Grossman W: Cardiac hypertrophy: useful adaptation or pathologic process. Am J Med 1980; 69: 576-84. [ Links ]
24. Gaasch WH: Left ventricular radius to wall thickness ratio. Am J Cardiol 1979; 43: 1189-94. [ Links ]
25. Guadalajara JF, Martínez C, Gutiérrez PE, Zamora C, Huerta D. et. al: Estudio de la función ventricular mediante la cuantificación de la relación grosor/radio (h/r) del ventrículo izquierdo en sujetos sanos. Arch Inst Cardiol Mex 1989; 59: 293-300. [ Links ]
26. Benjamín IJ, Schuster EH, Bulkley BH: Cardiac hypertrophy in idiopathic dilated congestive car-diomyopathy: a clinicopathologic study. Circulation 1981; 64:442-447. [ Links ]
27. Randhawa AK, Singal PK: Pressure Overload-induced cardiac hypertrophy with and without dilation. J Am Coll Cardiol 1992; 20: 1569-1575. [ Links ]
28. Carabello BA, Green LH, Grossman W, Cohn l, Koster K, Collins Jr. J: Hemodinamyc determinants of prognosis of aortic valve replacement in critical aortic stenosis and advanced congestive heart failure. Circulation 1980; 62: 42-48. [ Links ]
29. Escudero EM, Tufare AL, Ennis IL, Garciarena CD, Pinilla O A, Carranza VB: Análisis ecocardio gráfico del efecto de diferentes inhibidores del intercambiadorNa+/H+ sobre la estructura y función sistóli-ca del ventrículo izquierdo en ratas espontáneamente hipertensas. Medicina 2006; 66 (en prensa). [ Links ]
30. Guadalajara JF, González J, Bucio ER, Pérez P, Cué RJ: La cuantificación no invasiva del estrés parietal sistólico del ventrículo izquierdo en pacientes con insuficiencia cardíaca y su aplicación clínica. Arch Cardiol Mex 2007; 77: 120-129. [ Links ]
31. Gunther S, Grossman W: Determinants of ventricular function in pressure-overload hypertrophy in man. Circulation 1979; 59: 679-87. [ Links ]
32. Weber KT: Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 1989; 13: 1637-52. [ Links ]
33. Weber KT, Brilla CG: Pathological hypertrophy and cardiac interstitium: fibrosis and renin-angiotensin-aldosterone system. Circulation 1991; 83: 1849-65. [ Links ]
34. Weber KT, Janicki JS, Shroff SG, Clark WA: Pressure-overload hypertrophy: adaptive and pathologic remodeling of the myocardium. New Frontiers in Cardiovascular Therapy: Focus on Angiotensin Converting Enzyme Inhibition. Excerpta Medica an ElsevierC. 1989: 126-140. [ Links ]
35. Villardi B, Campbell SE, Hess OM, Mall G, Vassalli G, Weber KT, Krayenbuehl HP: Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease. J Am Coll Cardiol 1993; 22: 1477-84. [ Links ]
36. Lund O, Kristensen LH, Baandrup U, Hansen OK, Nielsen TT, Emmertsen K, et al: Myocardial structure as a determinant of pre-and postoperative ventricular function and long-term prognosis after valve replacement for aortic stenosis. Eur Heart J 1998; 19: 1099-1108. [ Links ]
37. Corin WJ, Monrad ES, Murakami T, Nonogi H, Hess OM, Krayenbuehl HP: The relationship of afterload to ejection performance in chronic mitral re gurgitation. Circulation 1987; 76: 59-67. [ Links ]
38. Guadalajara JF, Alexanderson E, Monobe F, Nieto S, Huerta D: La relación grosor/radio (h/r) del ventrículo izquierdo en la insuficiencia mitral pura. Arch Inst Cardiol Mex 1992; 62: 521-528. [ Links ]
39. Starling MR, Kirsh MM, Montgomery DG, Gross MD: Impaired left ventricular contractile function in patients with long term mitral regurgitation and normal ejection fraction. J Am Coll Cardiol 1993;22:239-250. [ Links ]
40. Wisenbaugh T, Spann JF, Carabello BA: Differences in myocardial performance and load between patients with similar amounts of chronic aortic versus chronic mitral regurgitation. J Am Coll Cardiol 1984; 3: 916-923. [ Links ]
41. Guadalajara JF, Galván Montiel, Noguera M, Alexanderson E, Cervantes JL, Huerta D: El mecanismo de remodelación en las sobrecargas de volumen del ventrículo izquierdo. Arch Inst Cardiol Mex 1995; 65: 217-28. [ Links ]
42. St. John Sutton MG, Shorpe N: Left ventricular remodeling after myocardial infarction, patho-physiology and therapy. Circulation 2000; 101: 2981-2988. [ Links ]
43. Guadalajara JF, Valenzuela F, Martínez C, Huerta D: La relación grosor/radio (h/r) en la miocardiopatía hipertrófica y dilatada. Arch Inst Cardiol Mex 1990; 60: 253-260. [ Links ]
44. Walther T, Shubert A, Falk V, Binner Ch, Walther C, Doll N, et al: Left ventricular reverse remodelin after surgical therapy for aortic stenosis: correlation to renin-angiotensin system gene expression. Circulation 2002; 106(Suppl I): I-23-I-26. [ Links ]
45. Weber KT, Brilla CG, Campbell SE: Regulatory mechanisms of myocardial hypertrophy and fibrosis: results of in vivo studies. Cardiology 1992; 81: 266-273. [ Links ]
46. Brilla CG, Janicky JS, Weber KT: Cardioreparative effects oflisinopril in rats genetic hypertension and left ventricular hypertrophy. Circulation 1991a; 83(5): 1771-9. [ Links ]
47. Brilla CG, Campbell SE, Matsubera L, Weber KT: Advanced hypertensive disease in SHR: Lisinopril mediated regression of myocardial fibrosis. Circulation 1992; 86 (Suppl I): 329 [ Links ](Abstract).

