










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.77 no.1 Ciudad de México ene./mar. 2007
Revisión de temas cardiológicos
Proteína C reactiva: aspectos cardiovasculares de una proteína de fase aguda
C–reactive protein: Cardiovascular issues of an acute–phase protein
Luis M Amezcua–Guerra,* Rashidi Springall del Villar,* Rafael Bojalil Parra*
* Departamento de Inmunología. INCICH.
Correspondencia:
Rafael Bojalil Parra.
Jefe, Departamento de Inmunología,
Instituto Nacional de Cardiología Ignacio Chávez.
(INCICH, Juan Badiano Núm. 1, Col. Sección XVI, Tlalpan, México D. F.).
Tel. 0155 5573 2911, ext. 1255. Fax. 0155 5573 0994
Recibido: 7 de marzo de 2006
Aceptado: 24 de octubre de 2006
Resumen
La proteína C reactiva (PCR) es una proteína inespecífica de fase aguda, utilizada como una medida de inflamación durante décadas. Recientemente se ha propuesto como un marcador de aterogénesis y como un predictor para el desarrollo de eventos cardiovasculares adversos a futuro. La PCR se une a lipoproteínas alteradas y facilita su remoción por los fagocitos, además de activar parcialmente el sistema del complemento. Los niveles elevados de PCR pueden producir efectos directos sobre las células vasculares, incluyendo inducción de citocinas y factores protrombóticos. Aunque inicialmente se había señalado una fuerte asociación entre los niveles de PCR y eventos cardiovasculares futuros, un meta–análisis reciente y varios estudios prospectivos han mostrado que esta asociación, si bien existe, pudiera ser más débil que lo descrito inicialmente. La terapia con estatinas en pacientes con enfermedad arterial coronaria ha mostrado reducir los desenlaces cardiovasculares adversos en asociación con una reducción de los niveles séricos de PCR, independientemente de su efecto sobre el perfil de lípidos.
Palabras clave: Proteína C reactiva. Enfermedad cardiovascular. Inflamación. Ateroesclerosis.
Summary
C–reactive protein (CRP) is a nonspecific acute phase protein that has been used as an inflammatory marker for decades. More recently, it has been proposed as a predictor of cardiovascular disease (myocardial infarction, stroke, peripheral artery disease and sudden heart death). Physiologic functions of CRP as an anti–inflammatory scavenger molecule have begun to emerge. CRP binds to damaged lipoproteins and facilitates their removal by phagocytes, partially activating the complement cascade. Increased levels of CRP may result in direct effects on vascular cells, including the induction of cytokines and prothrombotic factors. Although previous studies suggested a potent independent association of CRP levels with cardiac events, the strength of this association has been shown to be weaker than previously reported in a recent large meta–analysis and in prospective studies. Therapy with statins in patients with coronary artery disease has been found to reduce adverse outcomes in association with reductions of CRP levels, independently of their effects on the lipid profile.
Key words: C–reactive protein. Cardiovascular disease. Inflammation. Atherosclerosis.
Introducción
Dentro del espectro de las enfermedades con participación del sistema inmune, la cuantificación de la respuesta inflamatoria permite estratificar el estado basal de la enfermedad y evaluar su respuesta al tratamiento a través del tiempo. El tipo de parámetros bioquímicos empleados para este fin ha evolucionado a la par del conocimiento sobre los mecanismos fisiológicos y patogénicos que subyacen a la inflamación.
Considerando que la respuesta inflamatoria es un fenómeno dinámico integrado por múltiples procesos distintos e interdependientes, no es razonable esperar que una prueba de laboratorio los refleje a todos. Además, independientemente del tipo de estímulo, la respuesta inflamatoria utiliza de manera consistente mecanismos similares de acción, por lo que las pruebas de laboratorio que la cuantifican carecen de especificidad etiológica.
Fenómenos de fase aguda
Los fenómenos de fase aguda comprenden cambios bioquímicos inespecíficos en respuesta a diversas formas de daño tisular por infección, inflamación o neoplasia. Muchos elementos de esta respuesta parecen representar mecanismos de defensa tempranos de la inmunidad innata que preceden a la activación de la inmunidad adaptativa. De manera característica hay alteración en la síntesis de varias proteínas plasmáticas (Tabla I), con cambios en su concentración y en el tiempo de respuesta.

Tradicionalmente se ha empleado la velocidad de sedimentación globular (VSG) como la referencia para determinar inflamación; ésta es una medida indirecta de la concentración de diferentes proteínas plasmáticas (especialmente fibrinógeno) sintetizadas en abundancia durante una respuesta inflamatoria. Estas proteínas interactúan con la membrana de los eritrocitos, induciendo la formación de columnas eritrocitarias (rouleaux) más pesadas que los eritrocitos individuales, por lo que tienden a sedimentarse con mayor velocidad en el fondo de una columna de sangre.
La cuantificación directa de proteínas producidas durante los fenómenos de fase aguda es un método más fidedigno y confiable. De éstas, la proteína C reactiva (PCR) es la más difundida y la más accesible para el clínico. El advenimiento de inmunoensayos de alta sensibilidad para PCR en la década pasada, aumentó el interés en el estudio de esta proteína, no sólo como marcador de inflamación, sino como una molécula funcionalmente clave en las respuestas de la inmunidad innata. El concepto de que la PCR es también un mediador directo de diversos procesos patológicos llegó al demostrarse que los niveles séricos elevados de PCR (aún dentro de los parámetros considerados como normales) predicen el desarrollo de futuros eventos coronarios.
Así, en la práctica médica actual es indispensable un mejor conocimiento sobre los mecanismos fisiológicos y patogénicos relacionados con la PCR.
Descripción de la PCR
La PCR fue la primera proteína de fase aguda descrita y el nombre deriva de su capacidad para precipitar al polisacárido somático C del Streptococcus pneumoniae. La PCR forma parte de la inmunidad innata y su síntesis es inducida como respuesta al daño tisular por infecciones, inflamación o neoplasias. Es sintetizada por hepatocitos y células del endotelio vascular y su expresión está regulada por citocinas, particularmente por la interleucina 6 (IL–6) y, en menor grado, la interleucina 1 (IL–1) y el factor de necrosis tumoral α (TNF–α).1
La PCR pertenece a una familia de proteínas pentaméricas dependientes de calcio llamadas pentraxinas. Aunque la PCR se produce como monómero, la molécula funcional está compuesta por cinco subunidades polipeptídicas de 23 kDa idénticas asociadas de manera no covalente en una configuración anular con simetría cíclica. Las pentraxinas son proteínas que han subsistido a través de la evolución, con proteínas homologas entre especies filogenéticamente distantes. Sin embargo, hay grandes variaciones en la organización de las subunidades, en el ensamblaje proteico y en la cinética de la PCR entre especies, por lo que se deben extremar precauciones al extrapolar al humano los datos obtenidos en modelos animales.2
Mecanismos de acción propuestos de la PCR
La PCR se une con gran afinidad a una amplia variedad de ligandos tanto autólogos (lipoproteínas plasmáticas nativas y modificadas, membranas celulares dañadas, residuos de fosfatidilcolina, histonas, cromatina, ribonucleoproteínas pequeñas y células apoptóticas), como extrínsecos (glucanos, fosfolípidos y otros componentes somáticos y capsulares de bacterias, hongos y parásitos). Cuando la PCR está unida a ligandos macromoleculares es reconocida por C1q y activa la vía clásica del complemento; adicionalmente, provee sitios de unión para el factor H, regulando la amplificación de la vía alterna y a las convertasas de C5. Por otro lado, inhibe el ensamblaje de los componentes terminales del complemento (C5 – C9), atenuando la formación del complejo de ataque a la membrana y limitando la lisis celular por esta vía.3 Otros efectos de la PCR semejan algunas propiedades de la fracción cristalizable (Fe) de las inmunoglobulinas. Esta proteína es capaz de unir complejos inmunes y facilitar la depuración de detritus solubles y partículas apoptóticas, al ser reconocida por los receptores para la Fe de la IgG (FcγR) sobre los macrófagos activados.4
La capacidad de la PCR para activar el complemento y opsonizar partículas parece ser importante en la respuesta de la inmunidad innata frente a los patógenos. La ausencia de cualquier deficiencia congénita conocida de la PCR y su conservación filogenética sugieren que esta proteína debe tener gran importancia en la supervivencia de los individuos.
Cuando una célula en apoptosis es opsonizada y posteriormente fagocitada por macrófagos, induce la producción y liberación de diversas citocinas (como el factor de crecimiento transformante β), que inhiben el desarrollo de respuestas inmunológicas adaptativas. La PCR tiene la capacidad tanto de opsonizar células apoptóticas, como de desacoplar las proteínas del complejo de ataque a la membrana dependiente del complemento. Esto permite una mayor permanencia de las células apoptóticas antes de ser eliminadas, aunque facilitando su captación por fagocitos. Así, la PCR juega un papel preponderante en limitar la activación de respuestas de inmunidad adaptativa. Esto ha sido demostrado en modelos murinos carentes (knock–out) para el gen de la proteína amiloide sérica A (principal pentraxina en esa especie), los cuales desarrollan respuestas de autoinmunidad espontánea.5 Es interesante que los pacientes con lupus eritematoso generalizado, el prototipo de enfermedad mediado por complejos inmunes y anticuerpos autorreactivos, presenten una tasa muy baja de producción de PCR.6
Cinética de la PCR
La síntesis de novo de la PCR principia a las 6 horas después de iniciado el estímulo inflamatorio y alcanza su máximo a las 24–72 horas. Su vida media es relativamente corta (19 horas), pero su concentración plasmática es constante bajo cualquier condición y no se modifica con la ingestión de alimentos ni presenta variación circadiana, en contraste con las proteínas de la coagulación y otras de fase aguda. Una vez finalizado el estímulo de IL–6, la PCR regresa a valores normales al cabo de 7 días. Con esto, el índice de producción de la PCR es el único determinante de los niveles circulantes de la proteína, reflejando en forma directa la intensidad de los procesos patológicos que estimularon su síntesis.7
La concentración media de la PCR en donadores sanos es de 0.8 mg/L, pero después de un estímulo inductor, esta proteína puede incrementar su producción más de 10,000 veces. Los niveles séricos de PCR tienden a aumentar con la edad, probablemente como reflejo del incremento en la frecuencia de procesos inflamatorios subclínicos y de la cantidad de fenómenos apoptóticos. Se han detectado niveles séricos discretamente más elevados en mujeres que en varones.8
Al igual que la proteína amiloide sérica A (SAP), las características cinéticas de la PCR le proporcionan cualidades para considerarla como una proteína que refleja de manera fidedigna un fenómeno de fase aguda (Figura 1). Existe menos evidencia clínica sobre la utilidad de la SAP en comparación con la PCR y los ensayos para cuantificarla son poco accesibles y no estan estandarizados.
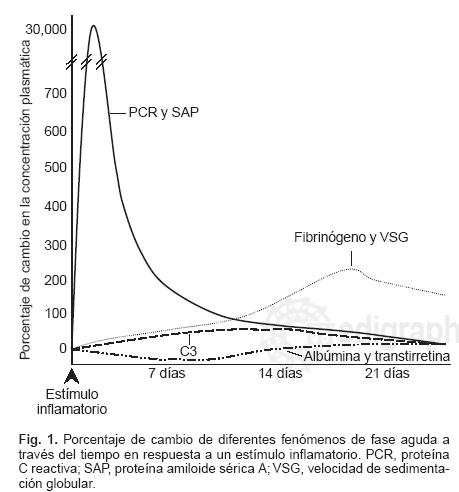
Interpretación de los niveles séricos de PCR
La síntesis de PCR depende de la concentración de mediadores inflamatorios producidos en el sitio de daño que llegan al hígado; por lo tanto, un valor normal de PCR no necesariamente indica ausencia de inflamación.
Empleando métodos de detección de alta sensibilidad, la distribución de PCR es muy similar entre géneros y grupos étnicos. Los valores de 0.3, 0.6, 1.5, 3.5 y 6.6 mg/L corresponden a los puntos de corte estimados para los percentiles 10, 25, 50, 75 y 90. Una forma alternativa más fácilmente asequible es el considerar valores de <1, 1 a 3 y >3 mg/L como grupos de bajo, moderado y alto riesgo para desarrollar eventos coronarios agudos a futuro.9
En general, cuando la PCR es < 10 mg/L traduce procesos inflamatorios leves como gingivitis, angina o ejercicio vigoroso. Elevaciones moderadas (10–100 mg/L) se encuentran en el infarto agudo del miocardio, la pancreatitis, las infecciones de mucosas (bronquitis, cistitis) y en la mayoría de las enfermedades reumáticas. Una concentración mayor de 100 mg/L se encuentra en las infecciones bacterianas agudas graves (como en la sepsis), traumatismos mayores (incluyendo quemaduras extensas) o vasculitis sistémica.10
Es importante aclarar que frecuentemente los valores de PCR se informan en mg/dL, por lo que habrá que tener especial cuidado sobre las unidades de medición al interpretar los resultados. Las ventajas de la PCR en comparación con otros indicadores de fase aguda, explican su uso cada vez más extendido (Tabla II). La utilidad de determinar los niveles séricos de PCR en la práctica clínica se presenta en la Tabla III. Para evitar posibles errores durante la determinación, se recomienda obtener dos mediciones de PCR con un intervalo de tiempo entre ellas y considerar el promedio de ambas.


Asociación entre enfermedad cardiovascular y PCR
Después de un infarto agudo del miocardio, los niveles séricos de la PCR se elevan rápidamente, reflejando la extensión de la necrosis. Los niveles máximos alcanzados a las 48 horas del evento agudo son útiles como factor pronóstico de la evolución de estos pacientes.11 Aunque esta elevada producción de PCR pudiera corresponder sólo a una respuesta de fase aguda típica a la muerte celular y a la infiltración inflamatoria subsiguientes, se ha demostrado que la PCR se deposita conjuntamente con fracciones activadas del complemento dentro de las zonas de infarto agudo,12 contribuyendo ambas a la gravedad de la lesión isquémica.13
Diversos estudios epidemiológicos han mostrado que los niveles séricos de PCR tienen valor predictivo para el desarrollo de síndromes coronarios agudos, eventos vasculares cerebrales, enfermedad arterial periférica y muerte súbita cardíaca.9
La relación existente entre los niveles básales de PCR y el riesgo de desarrollar eventos cardiovasculares ha sido consistente entre estudios. En muchos de ellos, mostrando independencia de la edad y de los factores de riesgo tradicionales como tabaquismo, niveles de colesterol, presión arterial y diabetes. Más aún, su valor predictivo se mantiene hasta por 20 años después de la primera determinación de PCR.14
Existe por lo menos una docena de estudios prospectivos donde se ha demostrado que al detectarse una concentración basal de PCR dentro de los cuartiles superiores, hay un riesgo relativo para desarrollar eventos cardiovasculares adversos de 2.0 a 4.0, en comparación con las concentraciones de PCR contenidas en los cuartiles inferiores (Figura 2).15–19
En un estudio prospectivo, Ridker y colaboradores15 demostraron que la PCR es un predictor de riesgo más potente que las lipoproteínas de baja densidad (LDL), con áreas bajo la curva (ROC) de 0.64 vs 0.60 y con un riesgo porcentual poblacional atribuible de 40 vs 19.
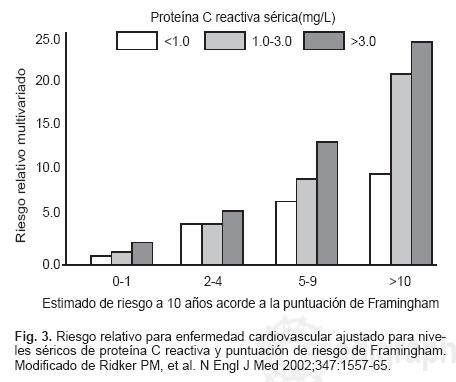
La PCR no muestra prácticamente ninguna correlación con los niveles de lípidos, por lo que no es posible predecir su valor a partir de la cuantificación del colesterol total, lipoproteínas de alta densidad (HDL) o LDL. La variación de la PCR adscrita a los niveles de LDL es menor de 5%.20 La PCR no suplanta el valor de las LDL en la predicción de riesgo cardiovascular, pero debe ser considerada como una prueba adjunta a la determinación de lípidos. El valor aditivo de la PCR al perfil de lípidos para la predicción de riesgo coronario ha mostrado que el presentar niveles de LDL < 130 mg/dL y de PCR > 3 mg/L, confiere un riesgo mayor que los niveles de LDL > 160 mg/dL y PCR < 1.0 mg/L. De la misma manera, los niveles de PCR impactan de manera significativa en todos los niveles de la evaluación de riesgo de Framingham (Figura 3).21
Recientemente se publicó un meta–análisis que incluyó 22 estudios prospectivos examinando a la PCR como predictor de eventos cardiovasculares futuros. Los 7,068 participantes incluidos para el estudio presentaron una edad media al ingreso de 57 años y un seguimiento de 12 años. Todos los estudios utilizaron determinaciones de alta sensibilidad y los resultados se ajustaron para al menos un factor de riesgo tradicional. Cuando se comparó el tercil superior contra el tercil inferior, la razón de momios ajustada fue de 1.58 (con IC 95% de 1.48 a 1.68).22 Otros estudios publicados desde este meta–análisis han mostrado riesgos relativos similares.23,24 Por ejemplo, en el Women's Health Study se encontró un riesgo relativo ajustado de 2.1, cuando se compararon las mujeres incluidas en el 10% más elevado de PCR en comparación con las incluidas en el 10% más bajo.25 El incremento en el riesgo cardiovascular adscrito a la PCR es consistente entre estudios. Sin embargo, después de ajustar los resultados con los factores de riesgo tradicionales, éste pareciera tener un valor más bajo al informado inicialmente.
Ateroesclerosis y PCR
Hay estudios recientes que sugieren que la PCR, además de reflejar la extensión del daño tisular post–infarto y de servir como un marcador serológico para la predicción de eventos coronarios agudos, contribuye directamente en la patogénesis, progresión y complicación de la enfermedad ateroesclerótica de manera directa. Por su capacidad de depositarse en la íntima de las arterias, la PCR provoca disfunción del endotelio, lo que facilita la activación, migración y alojamiento de los leucocitos en el interior de la íntima arterial. Esto contribuye a la formación de las lesiones vasculares que son la base del desarrollo de la aterosclerosis.26 Los efectos pro–inflamatorios y pro–aterogénicos de la PCR sobre las células endoteliales disminuyen la producción de óxido nítrico y prostaglandina I2 (previamente denominada prostaciclina), incrementan la secreción de IL–6, aumentan la expresión de moléculas de adhesión en la superficie endotelial (proteínas clave en el reclutamiento de monocitos y linfocitos T hacia los tejidos) y aumentan la secreción de quimiocinas (citocinas de bajo peso molecular con capacidad quimiotáctica). Todos estos son factores fundamentales en la migración de los leucocitos hacia la íntima de las arterias.27
Cuando las LDL alcanzan cierto umbral de concentración en la sangre, penetran al interior de la pared arterial donde son modificadas por procesos de oxidación. La PCR se une a estas lipoproteínas (tanto a las nativas como a las oxidadas) y facilita su fagocitosis e internalización por los macrófagos de la íntima arterial, promoviendo la formación de células espumosas (macrófagos con grandes cantidades de lipidos oxidados en su interior). La acumulación de células espumosas es determinante para la evolución patogénica de una placa arterial, haciéndola más propensa a erosionarse o romperse y liberando su contenido trombogénico. Esta propiedad protrombótica también se suma a la capacidad de la PCR para inducir la producción de factor tisular (iniciador de la cascada de coagulación) por los macrófagos activados. Ante el estímulo de la PCR, estas células incrementan la producción de especies reactivas del oxígeno y la síntesis de IL–1β, IL–6 y TNF–α.28 La PCR también induce un incremento en la síntesis de metaloproteasas, enzimas críticas que aceleran la degradación de diversos componentes de la matriz extracelular, causando debilitamiento de la capa fibrosa de las placas.29
Todos estos factores desestabilizan la placa de ateroma, haciéndola más vulnerable a la ruptura. Hay evidencia que sugiere acciones directas de la PCR en la inducción de apoptosis en las células del músculo liso de las arterias coronarias humanas (responsables de la síntesis de los componentes de la matriz extracelular), lo que favorece aún más la vulnerabilidad de la placa.30 Por lo tanto, es posible que las alteraciones en la concentración de la PCR sérica, además de reflejar la vulnerabilidad de las lesiones ateroesclerosas, participen directamente en su formación y ruptura.
Vigilancia de los niveles de PCR y opciones terapéuticas
La meta primaria de los programas de escrutinio cardiovascular es identificar individuos de alto riesgo en quienes se puedan modificar factores de riesgo tradicionales como tabaquismo, sedentarismo y dieta, así como lograr un control adecuado de la presión arterial, diabetes y dislipidemia. Una vez alcanzado este objetivo, la cuantificación en suero de la PCR se presenta como una herramienta adicional de predicción de riesgo útil para el clínico.
Actualmente no hay evidencia definitiva de que al disminuir los niveles de PCR se reduzca el riesgo cardiovascular. Sin embargo, muchas intervenciones reconocidas para disminuir el riesgo cardiovascular (pérdida de peso, dieta, ejercicio y suspensión de tabaquismo) se asocian con reducciones en las concentraciones séricas de PCR.
Varios agentes farmacológicos que reducen el riesgo cardiovascular modifican los niveles de PCR. El uso de diferentes estatinas (inhibidores de HMGCoA–reductasa) como pravastatina, lovastatina, cerivastatina, simvastatina y atorvastatina ha mostrado que, en promedio, disminuyen los niveles de PCR en un 15 a 25% a las 6 semanas de tratamiento. Los estudios CARE31 y PRINCE20 han mostrado que la magnitud en la disminución de LDL no guarda ninguna relación con la magnitud en la disminución de la PCR. Más aún, prácticamente la totalidad de los pacientes que reciben estatinas muestra disminución en los niveles de LDL, mientras que para la PCR se observa un claro patrón de pacientes con y sin respuesta. En el estudio CARE se mostró que el beneficio de la terapia con estatinas varió según las concentraciones de PCR, siendo la magnitud del beneficio de 55% para los que tienen la PCR elevada y de 30% en los que tienen PCR baja.32
Ridker y colaboradores han demostrado que los pacientes que alcanzan una cuantificación de PCR < 2 mg/L después del tratamiento con estatinas tienen mejores desenlaces cardiovasculares que aquellos con concentraciones > 2 mg/L, independientemente de la concentración sérica de LDL.33,34 Otros agentes hipolipemiantes como fibratos, niacina y gemfibrozil también han mostrado reducir los niveles de PCR, aunque su efecto para disminuir el riesgo cardiovascular no se conoce.9 La magnitud del efecto de la aspirina también parece guardar alguna relación con la determinación basal de PCR, pudiendo ser mayor en los pacientes con PCR elevada.35 De igual manera, se ha observado que las tiazolidendionas también modifican los niveles de PCR en pacientes con diabetes mellitus tipo 2, aunque su potencial beneficio cardiovascular se desconoce.36
Recientemente, Pepys y colaboradores37 han demostrado en un modelo de rata que al bloquear directamente la PCR con moléculas construidas por diseño (1,6–bis[fosfocolina]–hexano), se disminuye la extensión de la necrosis posterior al infarto de miocardio inducido. Este efecto protector es dependiente del sistema del complemento. Las implicaciones patogénicas y terapéuticas derivadas de este estudio son muy promisorias a mediano plazo.38
Conclusión
La PCR tiene múltiples efectos pro–inflamatorios y exacerba el daño tisular en diferentes patologías, llevando a una enfermedad más grave. Por otra parte, su deficiencia facilita el desarrollo de fenómenos de autoinmunidad, como el lupus eritematoso generalizado. La buena correlación de la concentración circulante de PCR con la gravedad, extensión y progresión de muchas patologías, es congruente con la idea de que esta proteína no es sólo un marcador sistémico de inflamación, sino que es un contribuyente directo en la patogénesis de diferentes enfermedades en las que participa la respuesta inmune innata.
Tanto para entender los mecanismos patogénicos reflejados en un valor elevado de PCR como para modificar el curso natural de sus potenciales complicaciones, es imperativo un nivel de conocimientos adecuado sobre esta proteína de fase aguda.
Referencias
1. Pepys MB, Baltz ML: Acute phase proteins with special reference to C–reactive protein and related proteins (pentraxins) and serum amyloid A protein. Adv Immunol 1983; 34: 141–212. [ Links ]
2. Baltz ML: Phylogenetic aspects of C–reactive protein and related proteins. Ann N Y Acad Sci 1982; 389: 49–75. [ Links ]
3. Gershov D, Kim S, Brot N, Elkon KB: C–reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implication for systemic autoimmunity. J Exp Med 2000; 192: 1353–63. [ Links ]
4. Pepys MB, Rowe IF, Baltz ML: C–reactive protein: binding, to lipids and lipoproteins. Int Rev Exp Pathol 1985; 27: 83–111. [ Links ]
5. Bickerstaff MCM: Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med 1999; 5: 694–7. [ Links ]
6. Vigushin DM, Pepys MB, Hawkins PN: Metabolic and scintigraphic studies of radioiodinated human C–reactive protein in health and disease. J Clin Invest 1993; 91: 1351–7. [ Links ]
7. Kushner I: Acute phase response. Clin Aspects Autoimmunity 1989; 3: 20–30. [ Links ]
8. Hutchinson WL, Koenig W, Frhlich M, Sund M, Lowe GDO, Pepys MB: Immunoradiometric assay of circulating C–reactive protein: age–related values in the adult general population. Clin Chem 2000; 46: 934–8. [ Links ]
9. Ridker PM: Clinical application of C–reactive protein for cardiovascular disease detection and prevention. Circulation 2003; 107: 363–9. [ Links ]
10. Ballou SP, Kushner I: Laboratory evaluation of inflammation. En Ruddy S, Harris ED, Sledge CB (Eds): Kelley's Lextbook of Rheumatology. Philadelphia, JB Saunders Company, 2001: 697–703. [ Links ]
11. De Beer FC, Hind CR, Fox KM, Allan RM, Maseri A, Pepys MB: Measurement of serum C–reactive protein concentration in myocardial ischaemia and infarction. Br Heart J 1982; 47: 239–43. [ Links ]
12. Lagrand WK, Niessen HW, Wolbink GJ, Jaspars LH, Visser CA, Verheuth FW, et al: C–reactive protein colocalizes –with complement in human hearts during acute myocardial infarction. Circulation 1997; 95: 97–103. [ Links ]
13. Griselli M, Herbert J, Hutchinsin WL, Laylor KM, Sohail M, Krausz L, et al: C–reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med 1999; 190: 1733–40. [ Links ]
14. Sakkinen P, Abbott RD, Curb JD, Rodriguez BL, Yano K, Lrac y RP: C–reactive protein and myocardial infarction. J Clin Epidemiol 2002; 55: 445–51. [ Links ]
15. Ridker PM, Rifai N, Rose L, Buring JE, Cook NR: Comparison of C–reactive protein and low–density lipoprotein cholesterol levels in the prediction of first cardiovascular event. N Engl J Med 2002; 347: 1557–65. [ Links ]
16. Lracy RP, Lemaitre RN, Psaty BM, Ivés DG, Evans RW, Cushman M, et al: Relationship of C–reactive protein to risk of cardiovascular disease in the elderly. Results from the Cardiovascular Health Study and the Rural Health Promotion Project. Arterioscler Thromb Vase Biol 1997; 17: 1121–7. [ Links ]
17. Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, et al: C–reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy–middle–aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation 1999; 99: 237–42. [ Links ]
18. Pradhan AD, Manson JE, Rossouw JE, Siscovick DS, Mouton CP, Rifai N, et al: Inflammatory biomarkers, hormone replacement therapy, and incident coronary heart disease: prospective analysis from the Women's Health Initiative observational study. JAMA 2002; 288: 980–7. [ Links ]
19. Mendall MA, Strachan DP, Butland BK, Ballam L, Morris J, Sweetnam PM, et al: C–reactive protein: relation to total mortality, cardiovascular mortality and cardiovascular risk factors in men. Eur Heart J 2000; 21: 1584–90. [ Links ]
20. Albert MA, Danielson E, Rifai N, Ridker PM: Effect of statin therapy on C–reactive protein levels: the Pravastatin Inflammation/CRP Evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286: 64–70. [ Links ]
21. Ridker PM, Glynn RJ, Hennekens CH: C–reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97: 2007–11. [ Links ]
22. Danesh J, Wheeler JG, Hirschfield GM, Eda S. Eiriksdottir G, Rumley A, et al: C–reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350: 1387–97. [ Links ]
23. Wilson PW, Nam BH, Pencina M, D'Agostino RB Sr, Benjamin EJ, O'Donnell CJ: C–reactive protein and risk of cardiovascular disease in men and women from the Framingham Heart Study. Arch Intern Med 2005; 165: 2473–8. [ Links ]
24. Ridker PM, Cook N: Clinical usefulness of very high and very low levels of C–reactive protein across the full range of Framingham Risk Scores. Circulation 2004; 109:1955–9. [ Links ]
25. Ridker PM, Rifai N, Cook NR, Bradwin G, Buring JE: Non–HDL cholesterol, apolipoproteins A–I and B100, standard lipid measures, lipid ratios, and CRP as risk factors for cardiovascular disease in women. JAMA 2005; 294: 326–33. [ Links ]
26. Zwaka TP, Hombach V, Torzewski J: C–reactive protein–mediated low–density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation 2001; 103: 1194–7. [ Links ]
27. Jialal I, Devaraj S, Venugopal SK: C–reactive protein: risk marker or mediator in atherothrombosis? Hypertension 2004; 44: 6. [ Links ]
28. Ballou SP, Lozanski G: Induction of inflammatory cytokine release from cultured human monocytes by C–reactive protein. Cytokine 1992; 4: 361–8. [ Links ]
29. Williams TN, Zhang CX, Game BA, He L, Huang Y: C–reactive protein stimulates MMP–1 expression in U937 histiocytes through FcgRII and extracellular signal–regulated kinase pathway: An implication of CRP involvement in plaque de stabilization. Arterioscler Thromb Vase Biol 2004; 24: 61–6. [ Links ]
30. Blaschke F, Bruemmer D, Yin F, Takata Y, Wang W, Fishbein MC, et al: C–reactive protein induces apoptosis in human coronary vascular smooth muscle cells. Circulation 2004; 110: 579–87. [ Links ]
31. Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E: Long–term effects of pravastatin on plasma concentration of C–reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100: 230–5. [ Links ]
32. Ridker PM, Rifai N, Pfeffer MA, Sacks FM, Moyela, Goldman S, et al: Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1998; 98: 8439–44. [ Links ]
33. Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, et al: Measurement of C–reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344: 1959–65. [ Links ]
34. Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al: C–reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352: 20–8. [ Links ]
35. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH: Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336: 973–9. [ Links ]
36. Haffner SM, Greenberg AS, Weston WM, Chen H, Williams K, Freed MI: Effect of rosiglitazone treatment on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. Circulation 2002; 106: 679–84. [ Links ]
37. Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al: Targeting C–reactive protein for the treatment of cardiovascular disease. Nature 2006; 440: 1217–21. [ Links ]
38. Kitsis RN, Jialal I: Limiting myocardial damage during acute myocardial infarction by inhibiting C–reactive protein. N Engl J Med 2006; 355: 513–5. [ Links ]

