










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.76 supl.4 Ciudad de México oct./dic. 2006
Investigación Clínica
De la reperfusión al post–acondicionamiento del miocardio con isquemia prolongada. ¿Nuevo paradigma terapéutico de los síndromes coronarios agudos con elevación del segmento ST?
De lo básico a lo clínico
Reperfusion and postcond in oning in acute st segment elevation myocardial infarction. A knew paradigm for the treatment of acute myocardial infarction. From bench to beside?
Eulo Lupi Herrera,* Jorge Gaspar,** Héctor González Pacheco,*** Carlos Martínez Sánchez,**** Gustavo Pastelín Hernández,***** Pastor Luna Ortiz,****** Edmundo Chávez Cosio*******
* Sub–director de la Sub–Dirección de Investigación Clínica. Del Instituto Nacional de Cardiología Ignacio Chávez.
** Jefe del Departamento de Hemodinámica. Del Instituto Nacional de Cardiología Ignacio Chávez.
*** Sub–Jefe del Departamento de Urgencias y Unidad Coronaria. Del Instituto Nacional de Cardiología Ignacio Chávez.
**** Jefe del Departamento de Urgencias y Unidad Coronaria. Del Instituto Nacional de Cardiología Ignacio Chávez.
***** Jefe del Departamento de Farmacología. Del Instituto Nacional de Cardiología Ignacio Chávez.
****** Ex–Jefe del Departamento de Anestesiología. Del Instituto Nacional de Cardiología Ignacio Chávez.
******* Sub–director de Investigación Básica y Tecnológica. Del Instituto Nacional de Cardiología Ignacio Chávez.
Correspondencia:
Eulo Lupi Herrera.
Instituto Nacional de Cardiología Ignacio Chávez
(INCICH, Juan Badiano Num. 1 Sección XVI, Tlalpan 14080, México, D.F.).
Resumen
En la génesis del daño inicial del miocardio que ha sufrido isquemia prolongada y posteriormente reperfusión, además del papel que puedan jugar los radicales libres derivados del oxígeno, se han involucrado: 1. el proceso de la re–energización. 2. la rápida normalización del pH tisular y 3. la regulación acelerada de la osmolalidad tisular. Causas que no son totalmente independientes y donde la ruptura mecánica del sarcolema parece ser el punto final del daño inmediato por la reperfusión. Al igual, la hipercontractura de las miofibrillas es muy probable que sea también una de las causas terminales mayores, misma que es producida por la re–energización de las células isquémicas, en las que se generan fuerzas contráctiles anormales, producto de la sobrecarga del calcio y de la fragilidad incrementada del citoesqueleto. Se sabe además, que la acidosis isquémica atenúa la activación de la hipercontractura. La rápida normalización del pH tisular, puede actuar como factor permisivo para la hipercontractura favorecida por la re–energización y a la vez contribuir a la mayor sobrecarga del calcio. En la condición de la reperfusión se produce edema, lo que se considera también una causa final del daño inmediato post– reperfusión. El mejor entendimiento de estos mecanismos que ocasionan el daño post–reperfusión nos ha permitido acercarnos hacia nuevas opciones terapéuticas, mismas que van encaminadas a interferir con tal deterioro. De manera reciente, se ha descrito en el terreno de la experimentación básica el fenómeno que se designa como "post–acondicionamiento". También, se ha consignado que el daño post–reperfusión se puede reducir de manera significativa al modificarse las condiciones hidromecánicas y la composición de lo reperfundido durante los primeros minutos del nuevo aporte sanguíneo al miocardio. Se conoce, que el post–acondicionamiento da la misma protección que el pre–acondicionamiento, pero a diferencia del último, el diseño del primero permite aplicarlo en la clínica en los enfermos con SICA C ESST. De manera naciente, se ha demostrado que el post–acondicionamiento existe en el hombre y que con su aplicación durante la realización de los PCI es factible proteger del deterioro por reperfusión a estas cohortes que sufren de isquemia aguda prolongada. Al limitar el daño del miocardio con una manipulación de naturaleza mecanicista, que luce simple en su implementación, la aplicación de ésta posiblemente resultará en beneficio clínico terapéutico. Más que nunca, en los terrenos de la isquemia prolongada y en el escenario del daño inmediato por reperfusión, es necesario ahondar en las investigaciones de estos estados patológicos, pues la aplicación del postacondicionamiento en el terreno de los SICA C ESST luce potencialmente relevante.
Palabras clave: Isquemia. Infarto agudo del miocardio. Reperfusión. Post–acondicionamiento.
Summary
After prolonged periods of ischemia and energy depletion, the ischemic myocardial cell can be jeopardized by specific causes within the reperfusion period. These causes can be viewed as unwanted aspects of the recovery process itself limiting its efficiency. Three potential initial causes of immediate reperfusion injury, aside from oxygen radicals, have been experimentally investigated in detail, and are briefly discussed: 1. re–energization; 2. rapid normalization of tissue pH; and 3. rapid normalization of tissue osmolality. These potential causes are not entirely independent. Understanding of the basic causes has opened novel perspectives for specific interference with these serious pathomechanisms. The experimental results obtained in the last years encourage the development of therapeutic approaches to reduce infarct size by specific measures applied during the early phase of reperfusion. In the clinical setting, reperfusion therapy for acute myocardial infarction (AMI) has shown to reduce mortality, yet it may also have deleterious effects, including myocardial necrosis and no–reflow. Almost two decades ago, great hope arose from the description of ischemic preconditioning. Unfortunately, ischemic preconditioning is not feasible in the clinical practice because the coronary artery is already occluded at the time of hospital admission of the AMI patient. Recently, in the dog model, a phenomenon called "post–conditioning" has been described. It has been reported previouly that reperfusion injury can be significantly reduced by modifying the conditions and the composition of the initial reperfusate. Whereas preconditioning is triggered by brief episodes of ischemia–reperfusion performed just before a prolonged coronary artery occlusion, postconditioning is induced by a comparable sequence of reversible ischemia–reperfusion, but it is applied "just after the prolonged" ischemic insult. Protection afforded by postconditioning is as potent as that provided by preconditioning. Unlike preconditioning, the experimental design of postconditioning allows direct application in the clinical practice, especially during PTCA. It has been reported very recently, that postconditioning patients with ST segment elevation AMI, during coronary angioplasty protects the human heart in this clinical scenario. Obtaining such a beneficial effect by a simple manipulation of reperfusion is of major potential clinical interest. Now more than ever, mechanistic and pharmacological research in the field of reperfusion injury appears to be necessary and clinically relevant.
Key words: Ischemia. Myocardial infarction. Reperfusion. Postconditioning.
Introducción
El único método terapéutico que ha sido capaz de limitar el tamaño del infarto agudo del miocardio (IAM), es la reperfusión oportuna y exitosa de la arteria responsable de éste (ARI), con lo que se ha logrado reducir la morbi–mortalidad de los síndromes coronarios agudos con elevación del segmento ST (SICA C ESST).1–4 Aunque el pronóstico del IAM ha mejorado, aún representa una causa mayor de mortalidad y del desarrollo de insuficiencia cardíaca, tanto en los países desarrollados como en los que están en vías de industrialización. Sabemos que el tamaño del IAM es una determinante importante de la sobrevivencia a corto y a largo plazo,5,6 y que la reperfusión se acompaña de situaciones fisiopatológicas que no se consideran favorables para obtener un resultado final óptimo, lo que incluye el desencadenar mayor proporción de miocardio afectado o la presencia del llamado fenómeno de no – reflujo.7,8En general este proceso deletéreo se conoce como "daño por reperfusión". Por lo tanto, es una condición patológica que está asociada a éste, pero que no ha acontecido durante el período de isquemia que le precede y que a la vez pudiera ser atenuada mediante alguna acción al momento de restablecer la permeabilidad de la ARI. El daño post – reperfusión, se considera un problema mayor a resolver, de manera particular después de que se ha reperfundido de forma oportuna el miocardio que ha sufrido isquemia prolongada.8,9Es interesante señalar, que la existencia de esta condición fue cuestionada en el pasado por investigadores muy connotados en el tema como Kloner RA,9 y por Ferrari R y Hearse DJ.10Esta duda, ha tenido su origen en la problemática que resulta seguir el curso de la necrosis en el tiempo, tanto en los modelos animales como en el hombre que sufre de isquemia aguda prolongada. Para resolver esta situación académica adversa, los investigadores han utilizado un camino experimental ideológico opuesto y éste consiste en "modificar la naturaleza de la reperfusión" y cuantificar si la magnitud de la necrosis se ve finalmente reducida.8 Por otro lado, el "pre–acondicionamiento isquémico" del miocardio se conoce desde el año de 1996, cuando Murry CE, Jennings RB y Reimer KA,11 establecieron el concepto revolucionario que creó el saber que episodios repetidos y breves de isquemia protegen, en vez de acentuar el daño miocárdico final. Así, reducciones de hasta un 75.% del deterioro del miocardio, se logran alcanzar en el animal de experimentación, mediante esta maniobra de pre–acondicionamiento. Sin embargo, el mayor problema clínico y práctico con el pre–acondicionamiento isquémico, radica en que la protección que brinda éste tendría que aplicarse antes de que aconteciera la oclusión de la ARI, es decir el de un IAM no anunciado. En el año del 2003 Zhao ZQ et al,12 hacen una comunicación muy relevante, cuando demuestran que la aplicación del mecanismo de postacondicionamiento del miocardio una vez realizada la permeabilidad de la ARI reduce el "daño post – reperfusión", al documentar que la liberación enzimática (utilizada como índice de afección miocárdica irreversible) es abatida de manera significativa. Esta valiosa indagación, ha sido confirmada por otros investigadores en los escenarios experimentales que analizan el daño letal post–reperfusión y hoy día por Staat P et al,13 en los SICA C ESST. En esta revisión el primer objetivo será, comentar los conceptos actuales del daño post–reperfusión, en segundo término los relacionados con el post–acondicionamiento y en tercera circunstancia los aspectos del posible impacto que tenga éste al aplicarlo en la reperfusión de la ARI en la clínica. Se irá entonces, de los hallazgos experimentales básicos documentados en el laboratorio del daño post– reperfusión, al mecanismo de post–acondicionamiento del miocardio reperfundido, analizando el posible impacto que tenga en la cabecera del enfermo con SICA C E SST.
El daño letal post–reperfusión
Éste se define como el deterioro causado por la restauración del flujo coronario que acontece después de un episodio isquémico prolongado que ha dado cierta magnitud de daño a las células miocárdicas. Para que se acepte el concepto, la isquemia previa no debe de haber ocasionado un daño final irreversible o total. Por lo tanto, hay que incluir en el corolario: que las alteraciones parciales de la células del miocardio son un pre–requisito de la isquemia previa, para que de manera ulterior pueda acontecer "el daño post– reperfusión" (Fig. 1). De acuerdo a Piper HM et al,8 es técnicamente imposible evaluar las manifestaciones del insulto miocárdico irreversible en la misma pieza del miocardio antes y después de la reperfusión y tampoco distinguir en el análisis si la muerte celular fue producto de la isquemia previa o de la maniobra de la reperfusión. Ellos señalan, que la aplicación de este criterio directo no es permisible, pero sí existe uno de tenor indirecto que ha validado la presencia del deterioro letal post–reperfusión. Éste se basa en el concepto de modificar las condiciones de la reperfusión, lo que finalmente cambiaría la posibilidad de prevenir la muerte celular que de manera natural acontecería en el binomio isquemia – reperfusión. Aunque el razonamiento antes señalado luce simple, debemos de estar conscientes de que aunque se realicen maniobras que vayan encaminadas a modificar la reperfusión, el no encontrarlo no niega la existencia del daño ocasionado por la reperfusión. Otros aspectos que merecen comentarse es que, en algunos de los modelos experimentales el punto final de la muerte celular se ha analizado a las horas de haberse efectuado la reperfusión, lo que no está ligado necesariamente al proceso de protección de la nueva irrigación, lo que sí parece estar dirigido a la valoración de la reducción final del tamaño del infarto.9 Puesto que el proceso de acondicionamiento post–reperfusión ocurre en los primeros "segundos a minutos" de haber alcanzado la permeabilidad de la ARI, esta revisión del daño post–reperfusión se concentrará principalmente en los mecanismos por los que acontece la muerte celular, es decir en los primeros "minutos a las primeras horas" en la que se ha consumado ésta.10–14
Por lo que en el escenario clínico el daño del miocardio producto del complejo isquemia –reperfusión debe de considerarse como una sola entidad en la práctica cardiológica. El perjuicio que ocasiona la isquemia – reperfusión del miocardio es compleja e igual perturba a las estructuras celulares de los vasos como a los cardiomiocitos. Sin embargo, finalmente también se debe de tener en cuenta que la afección letal "inmediata" como la "tardía" pueden hacer sufrir al cardiomiocito al activarse los neutrófilos polimórficos y producir como consecuencia inducción de la apoptosis en el miocardio reperfundido.
Las causas del deterioro letal inmediato post–reperfusión
En un número de ensayos experimentales los efectos de los antioxidantes se han investigado en este escenario. Partiendo de la hipótesis que señala que durante la reperfusión se liberan o se producen radicales libres derivados del oxígeno (ROS) y que éstos son la causa fundamental de la afección post–reperfusión.8 La presunción anterior, se ha fundamentado en dos hechos bien documentados: 1. que los ROS en efecto si se promueven durante la reperfusión y 2. que la administración exógena de los mismos tienen efectos negativos en los sistemas celulares y en los sub–celulares. De acuerdo a Piper HM, García–Dorado D y Ovize M,8 la presunción anterior se ha visto limitada por los siguientes hechos: que no existe en el mecanismo un vínculo estrecho que explique el daño que ocasionan los ROS con la hiper–contractura de las miofibrillas y la muerte celular (Fig. 1). Otro factor que ofreció confusión en la mencionada hipótesis fue el establecer el mecanismo productor entre el aturdimiento y el desarrollo de la apoptosis post–reperfusión. La mezcla de estas razones, hizo inclusive llegar a negar la existencia del daño post–reperfusión y que los ROS no eran indispensables para su producción. En contraste con estos aspectos, sí hay certidumbre contundente que los ROS juegan un papel importante en la génesis del aturdimiento del miocardio y que los antioxidantes alivian éste.14 Sin embargo, hoy día se conoce que los ROS no son los únicos candidatos para la producción del daño letal post–reperfusión y que sí hay otros caminos que intervienen en su génesis. Además del papel que puedan jugar los ROS, se han involucrado en el perjuicio inicial post–reperfusión: 1. el proceso de la re–energización. 2. la rápida normalización del pH tisular y 3. la regulación vertiginosa de la osmolalidad tisular. Causas que no son totalmente independientes, donde la ruptura mecánica del sarcolema parece el punto final del daño inmediato por la reperfusión. Al igual, la hiper–contractura de las miofibrillas es muy probable que sea una de las fuentes terminales mayores, misma que es producida por la re–energización de las células isquémicas en donde se generan fuerzas contráctiles anormales, producto de la sobrecarga del calcio y de la fragilidad incrementada del citoesqueleto. Se conoce además, que la acidosis de naturaleza isquémica atenúa la activación de la hiper–contractura. La rápida normalización del pH tisular, puede actuar como factor permisivo para la hiper–contractura favorecida por la re–energización y a la vez contribuir a la mayor sobrecarga del calcio. En la condición de la reperfusión se produce edema, lo que se considera también una causa final del daño letal inmediato post–reperfusión. El edema se origina como producto de una normalización muy rápida de la osmolalidad extracelular dejando el fluido intracelular hiper–osmolar.14–20
El papel de la re–energización
Hearse DJ et al,15 en el año de 1973 demostraron que en el miocardio depletado de oxígeno y nuevamente oxigenado, el daño de éste se caracteriza por hiper–contractura miofibrilar y por la disrupción del sarcolema. Consideración que ha sido avalada por Ganóte CE et al,16,17quien ha demostrado que se debe a la nueva producción de energía al acontecer la re–oxigenación del miocardio previamente sometido a la isquemia prolongada. Este fenómeno de daño celular del miocardio producido de manera inmediata por la re–energización se ha designado como: la "paradoja del oxígeno".15 Sin embargo, a pesar de existir estas aportaciones de la investigación, se ha mantenido como una pregunta vigente si la paradoja del oxígeno representa un genuino insulto de la re–oxigenación o es una representación espectacular por el daño que ya ha acontecido durante el período de la depleción del oxígeno. La presencia de las bandas de contracción en el tejido miocárdico infartado es un indicador histológico del daño de la paradoja del oxígeno en el lecho miocárdico que se ha sometido a isquemia–reperfusión. El análisis histológico ha demostrado que cuando la reperfusión se ha realizado de manera oportuna para llegar a salvar tejido miocárdico, los infartos están compuestos principalmente por bandas de contracción, lo que refleja la hiper–contractura de los miocitos; además hay datos que dan soporte a que el mencionado proceso contráctil anómalo acontece durante los "primeros minutos del reflujo coronario".9,16,17 Aunque las bandas de contracción se pueden llegar a observar en ausencia de necrosis, como consecuencia de la aplicación de artefactos durante las tomas de biopsias, en el escenario de la reperfusión la hiper–contractura de los miocitos acontece en asociación con la necrosis. Hecho que indica que opuesto a lo que sucede en tratándose de miocitos aislados, la hiper–contractura inducida por el reflujo se ve coligada a disrupción del sarcolema y a la muerte celular (Fig. 1). Los estudios realizados en cardiomiocitos aislados han demostrado que la paradoja del oxígeno es en realidad una ofensa producto del proceso de la re–oxigenación cuyo mecanismo acontece dentro del cardiomiocito. La re–energización causa daño por reperfusión al provocarse la hiper–contractura y el mecanismo expresado de manera simplista es el siguiente: después de haber existido un estado en el cual está depletada la energía, la concentración del calcio del citosol queda dramáticamente elevada. El proceso de la re–energización de la célula miocárdica es posible, al suplirse nuevamente de oxígeno la mitocondria, en este momento dos causas se activan de manera simultánea, la energía suministrada por la bomba de cationes inicia el recobro del balance celular de éstos y al reasumir la energía a los elementos miofibrilares se inicia la actividad contráctil.8,9,15–20 In vitro, es factible proteger a las células miocárdicas reoxigenadas de los estados hiper–contráctiles al abatir los movimientos oscilatorios del calcio, reduciendo las concentraciones elevadas de este ion. Proceso que se puede alcanzar mediante el bloqueo específico de la captura del calcio en el retículo sarcoplasma con ácido ciclopiazónico o con rianodina. Es interesante señalar que la acción del anestésico volátil halotano también puede inhibir la función del retículo sarcoplásmico y por lo tanto brindar protección en este sentido. El halotano aplicado en el momento de la re–oxigenación, ha demostrado ser protector contra la hiper–contractura y el daño post–reperfusión de los cardiomiocitos aislados, en corazones sometidos a la hipoxia y posteriormente re–oxigenados y en el miocardio in vivo sometido a la isquemia –reperfusión.21–25
El impacto de la normalización rápida del pH tisular
El pH de los cardiomiocitos que se han sometido a reperfusión tiene profunda influencia sobre el proceso en la aparición de la desproporción que se ha notado de la contractura. Después de una isquemia prolongada, el pH del citosol desciende de manera significativa como consecuencia de haberse establecido el metabolismo anaeróbico el que aumenta la degradación del ATP y produce un exceso de la concentración de los iones de hidrógeno (Fig. 1). Esto provoca acidificación del medio tanto en el espacio intra como en el extracelular. Al acontecer la reperfusión, el pH del espacio intersticial se normaliza nuevamente lo que genera un gradiente entre el del citosol, que contiene aún elevadas cantidades del ion H+ y el intersticio donde la concentración del ion H+ se ha ya regularizado. Este proceso tiene dos consecuencias: 1. la acidosis intracelular es velozmente reducida. 2. la rápida extrusión del exceso de protones de las células remueve el efecto potencialmente protector del agente y la activación del intercambio Na+/H+ causa un influjo neto del Na+ al citosol.26–28Dependiendo de la habilidad de la bomba de sodio para remover el exceso de éste, se establece un segundo mecanismo del intercambio Na+/Ca2+, transportando el Na+ en una dirección opuesta a la del Ca2+. Este proceso puede favorecer la carga elevada pre–existente del Ca2+ en las células. La remoción rápida del H+ y la captación secundaria del calcio favorecen el desarrollo de la hiper–contractura si las células miocárdicas sometidas a la isquemia – reperfusión son capaces de restablecer su balance ácido–base intracelular.28,29Se ha demostrado in vitro, que el efecto continuado de la acidosis dentro de la célula durante la fase temprana de la re–oxigenación, protege a las células miocárdicas, haciendo que éstas no desarrollen la hiper–contractura en esta fase. Mas es de hacerse notar que, para el miocardio in vivo y en estas circunstancias de la reperfusión la situación es menos clara. Así mismo y de manera desafortunada hasta la fecha, no se han descrito en los ámbitos de la investigación básica inhibidores netamente específicos de estos mecanismos.9,26–30
El impacto de la rápida normalización de la osmolalidad tisular
Una de las principales causas del paso de agua a las células del miocardio sometidas a la isquemia – reperfusión es la sobrecarga de Na+ en el citosol. El intercambio de Na+/H+juega un papel mayor en la regulación del volumen celular. En el caso del miocardio isquémico los productos finales del metabolismo anaeróbico se ven acumulados, los que aumentan la carga osmótica en el espacio intra y en el extra–celular. 29,30 Si durante la reperfusión, el exceso de moléculas osmóticas activas se lavan, se genera un gradiente de esta naturaleza entre los espacios intra y extra – celular. Al existir una mayor captación celular de agua, con el consecuente incremento de la presión intracelular se produce un estiramiento mecánico del sarcolema con un aumento de la fragilidad mecánica, misma que ya pre–existía durante el período de depleción de la energía. Como es el caso de la hiper–contractura, el edema de por sí habitualmente no es capaz de romper el sarcolema. Situación que se ha demostrado en condiciones de cardiomiocitos aislados sometidos al estrés osmótico en condiciones de oxigenación normal donde el sarcolema mantiene su integridad.31–33 En cardiomiocitos aislados, el estrés induce oncosis sólo sí existe hiper–contractura y si de manera prolongada se han sometido a la deprivación prolongada de energía. De tal manera que, la combinación de estos factores aumentan la fragilidad y hacen que la célula sea más susceptible al efecto del estrés osmótico. Los estudios realizados por investigadores como Kloner RA et al,34 con reperfusiones altamente hiper–osmóticas indican que, el atenuar el efecto adicional mecánico del estrés osmótico impuesto por el edema limita la zona de la necrosis miocárdica durante la reperfusión. En cuanto al mecanismo de la fragilidad del sarcolema secundario a la deprivación de la energía aún no se conoce bien en todo su detalle. Sin embargo, las alteraciones en la composición de los lípidos de la membrana, los cambios en las proteínas del sarcolema o las modificaciones entre el sarcolema y el citoesqueleto pueden jugar algún papel. También se ha demostrado que la isquemia induce fragilidad del sarcolema y que ésta es agravada durante los primeros momentos de la reperfusión.8 En todo este escenario es de consignarse que los ROS han resultado ser un factor secundario en la génesis del daño post–reperfusión.
La interacción célula – célula
Varias líneas de investigación han demostrado que la re–oxigenación induce hiper–contractura y la ruptura del sarcolema y que a la vez éstas son influenciadas por la interacción célula–célula.21,26,29,30
Las observaciones histológicas, han indicado que las bandas de contracción de necrosis que son producto de las oclusiones temporales de las arterias y que son seguidas de reperfusión se componen de miocitos hiper–contraídos que están conectados unos a otros y que llegan a formar un manto de continuidad, lo que resulta en un complejo geométrico, hecho que no puede explicarse sólo con fundamento a gradientes de flujo o a la distribución del daño microvascular (Fig. 1). Estudios de simulación computational, han indicado que cierta interacción de célula a célula debe de tomarse en cuenta para explicar este panorama histológico anormal, ya que en ausencia de tal interdependencia, los miocitos con hiper–contractura deberían representar áreas aisladas en la zona de riesgo en vez de formar un manto de continuidad.8 En este sentido, se ha postulado que en relación a esta inter–acción de célula a célula debe de tener una naturaleza mecánica, producto de fuerzas intercambiables impuestas por las estrechas uniones inter–celulares, las que desgarran los sarcolemas de los miocitos con hiper–contractura durante la reperfusión in situ, lo que a la vez deteriora el sarcolema de estos elementos adyacentes. Sin embargo, también se acepta que la interacción entre las células puede ser de naturaleza bioquímica.35–37Estudios recientes en cardiomiocitos aislados, han demostrado que la hiper–contractura inducida por la ruptura del sarcolema o por la micro inyección de calcio que produce la hiper–contractura de las células adyacentes.8,35–37La administración intracoronaria de heptanol durante los primeros minutos de la reperfusión se ha visto que abate significativamente el tamaño del infarto del corazón en el animal de experimentación (como es en el caso del cerdo) que es sometido a una oclusión coronaria transitoria. Resultados que apoyan el hecho hipotético que la transmisión célula – célula producto de la hiper–contractura, puede ser una causa por la que se extienda la necrosis en el momento del daño por la reperfusión.
La apoptosis y la necrosis de aparición tardía
Algunas intervenciones con miras a modificar las consecuencias de la reperfusión pueden aparentemente reducir la extensión del daño tisular cuando se investiga esta posibilidad de manera temprana; sin embargo, lo único que puede acontecer es que en realidad retrasen la aparición del daño final irreversible del miocardio. Si esta es la situación, tal manipulación no brinda una protección real en la reperfusión. De acuerdo a Piper HM et al,8 este aspecto debe claramente de distinguirse de las causas que se hacen presentes no en el momento de la reperfusión sino de manera mucho mas tardía, como es la que dan los neutrófilos activados que producen invasión tisular con su respectivo perjuicio. Dentro de este mismo grupo de alteraciones tardías, está la posibilidad de si el miocardio sujeto a reperfusión puede sufrir el proceso de la apoptosis aunque sea protegido de manera inmediata del daño. A diferencia de lo que acontece para la muerte de la célula, que es una consecuencia de un grave detrimento estructural y que no es mediada por trascripción, en cambio la apoptosis sí lo es y acontece en células con deterioro moderado como consecuencia de la acción de las citoquinas. En estudios recientes, se ha comunicado que en el transcurso de la isquemia – reperfusión y en particular en los bordes isquémicos se ha demostrado la presencia de la apoptosis. Observaciones que han dado lugar al cuestionamiento si la reperfusión del miocardio isquémico es seguido por la apoptosis, lo que finalmente limitaría el efecto benéfico que da la misma.37–39De acuerdo a la opinión de Piper HM et al,8 en la contribución del proceso de apoptosis aún no se han establecido los factores que inician éste en el miocardio que se ha sometido a la isquemia –reperfusión. Tampoco se ha puntualizado si los generadores de la apoptosis ocurren en el período de isquemia o es en el lapso de la reperfusión. De manera sintetizada podemos señalar que una vez sometido el miocardio a una isquemia "prolongada" y al acontecer la reperfusión, el daño letal inmediato por la nueva irrigación parece producto de la: 1. rápida normalización del pH. 2. del nuevo aporte de energía 3. del aumento de la fuerza de contracción, lo que produce hiper–contractura. 4. de la disrupción del sarcolema y 5. posiblemente de la transmisión de daño célula–célula.8,21,22,26–32,34–39 El conocer y tener presente estas causas básicas, las que no lucen deseables en el proceso de recuperación del miocardio, permiten entender mejor la limitación del recurso terapéutico de la reperfusión en los SICA C ESST.
El post–acondicionamiento del miocardio
Aunque la reperfusión salva parte del miocardio que se ha sometido a la isquemia prolongada y que ultimadamente éste moriría sin ella, el restaurar el flujo coronario, como ya se ha señalado puede tener implícita la potencialidad de exacerbar el daño ya presente y que es consecuencia de la isquemia. De tal manera que, con la maniobra antes mencionada, de cierta manera se opaca el beneficio que se pretende alcanzar con la cirugía de revascularización coronaria (CRVC), con los procedimientos coronarios intervencionistas (PCI) o con la fibrinólisis. Las estrategias iniciales de ofrecer protección o de atenuar el deterioro post–reperfusión del miocardio han sido un punto presente siempre desde que se aplican los métodos quirúrgicos en este órgano. Las manipulaciones miocardio–protectoras en este escenario se han centrado en modificar las diversas condiciones de la reperfusión como han sido la temperatura, la presión de reperfusión y la aplicación de las llamadas infusiones cardiopléjicas (pH, osmolalidad, utilización de substratos como la glucosa, aminoácidos y algunos medicamentos), mismas que modifican la composición del fluido reperfundido, situaciones que después de un cierto período de isquemia administradas a baja presión han logrado limitar el tamaño del infarto o hacer menos probable la presencia del llamado corazón de piedra.40,41 En forma muy sucinta a lo antes mencionado, es que se modifican las condiciones o los compuestos de la reperfusión, mismas que se han usado por más de 30 años y que sí han demostrado que abaten algunos aspectos deletéreos de ésta en la CRVC. Así todas las estrategias iniciadas antes o al momento del reflujo coronario y que incluyen la utilización de fármacos se pueden considerar como medidas que van encaminadas a modificar la reperfusión. Okamoto F et al,42 en el año de 1986 hicieron una observación que se puede considerar pionera y muy relevante, misma que ha sido validada por otros investigadores. Ésta se refiere al hecho de haber notado que el reiniciar el flujo coronario a una presión baja o atenuada (inicio lento del reflujo) reduce el tamaño del infarto. Esta nueva promoción gentil de la reperfusión arterial coronaria minimiza el área del infarto, el edema de la zona en riesgo y al parecer la posibilidad de que aparezca el déficit circulatorio que conocemos como fenómeno de no – reflujo. El "post–acondicionamiento" del miocardio, es un conocimiento y una estrategia que tuvo su origen en una idea paralela al concepto del pre–acondicionamiento del miocardio. Los investigadores responsables de ello fueron Zhao – QZ et al (Figs. 2, 3 y 4).12Ellos desplazaron esta estrategia irrigatoria previa a la isquemia al inicio de la reperfusión, lo que sensiblemente modificó favorablemente esta última. Es de mencionarse que en los ensayos experimentales realizados 20 años atrás, en donde se aplicaban ciclos alternos de cinco minutos de reperfusión y cinco de oclusión coronaria, que precedían a la restauración completa del flujo coronario, no fueron capaces de expresar de manera final una reducción del tamaño del infarto.43 Con fundamento en estas observaciones experimentales que fueron lamentablemente negativas, el concepto de postacondicionamiento del miocardio en el contexto del daño post–reperfusión se consideró concluida y permaneció olvidada por más de 10 años en el terreno de la investigación básica. De acuerdo a Viten–Johansen J et al,43 la razón de tal interpretación se debió a que los protocolos iniciales encaminados a este análisis de la isquemia al parecer fueron en general en su diseño subóptimos. Afortunadamente y debido al mejor entendimiento y a la maduración académica del fenómeno del daño post–reperfusión fue que se apreció lo importante que es considerar el "tiempo en segundos a minutos" en este contexto patológico del miocardio con isquemia prolongada. Gracias a los avances de la medicina experimental básica, se documentó que muchas de las alteraciones acontecían en este escenario en cuestión de segundos a minutos una vez provocada la reperfusión. De manera notable se ha llegado a documentar que numerosas reacciones físicas y mecanismos bioquímicos acontecen muy rápidamente al instalarse la reperfusión. Así se generan en este lapso los ROS, la activación de los neutróñlos y su adherencia al endotelio vascular coronario, el daño continuado del mismo a medida que progresa la reperfusión y la homeostasis anormal del calcio que daña las estructuras celulares más íntimas.43–47 Estos conocimientos nacientes, en la acción tan rápida de estos generadores del daño post–reperfusión, han hecho resurgir el concepto del postacondicionamiento y han permitido modificar la secuencia experimental que iba de varios minutos en los ciclos de oclusión – reperfusión, a la modalidad que va ahora de "segundos a un minuto" (Fig. 2). El establecer esta secuencia en este lapso ha demostrado que se atenúa el detrimento en los cardiomiocitos, se reduce el tamaño del infarto, la apoptosis y también el menoscabo del endotelio vascular coronario. El post–acondicionamiento mitiga la acción de muchos provocadores del daño post–reperfusión que abarcan los oxidantes, las citocinas pro–inflamatorias, los neutrófilos, los reguladores de la pro–apoptosis, lo que comprende un amplio espectro terapéutico que muchas de las medicaciones que se pretenden usar en este escenario para dar protección no la poseen o no se les ha distinguido.46–48 La maniobra de post–acondicionamiento del miocardio ha demostrado ser efectiva en todas las especies sometidas a ella, con la excepción del cerdo y con el hecho muy notable que sí lo es en el humano.13,49
Los estudios experimentales
Como ya se ha señalado, la reperfusión oportuna del miocardio salva al tejido que se ha sometido a isquemia prolongada y también sostiene que la súbita restauración del flujo coronario, hasta cierto punto de una manera paradójica, exagera el daño que no ha estado presente al momento de haberse concluido el período de isquemia prolongada. La magnitud de este deterioro miocárdico se ha visto que se puede minimizar modificando en principio las condiciones hidrodinámicas del torrente vascular arterial, es decir la presión de perfusión y la magnitud en el aporte del flujo coronario. La reperfusión abrupta, en los primeros minutos efectuada a presión sistémica puede alterar las fuerzas de Starling en el territorio microvascular y así favorecer el curso de los líquidos hacia el espacio intersticial, potencialmente precipitar la compresión microvascular y crear el fenómeno de no–reflujo. Sin embargo, algunos estudios han demostrado resultados contradictorios al realizar la reperfusión coronaria hecha aparentemente controlada. Farber N et al,50 no lograron definir una acción favorable en la función miocárdica regional entre los cero y 10 minutos en la fase de reperfusión y es más, determinaron un deterioro del acortamiento sistólico a los 180 minutos de haberse realizado ésta. Granate JE et al,51,52 y Yamazaki S y colaboradores,53 no consignaron modificaciones favorables en cuanto al tamaño del IAM, habiéndose realizado oclusiones coronarias de una a tres horas seguidas de un largo período de reperfusión. En contraste con la información previa, otros estudios sí han asignado resultados positivos al haberse realizado la reperfusión con un cuidadoso control hidrodinámico. Hori M et al,54 en el año de 1991 demostraron de manera precisa que la reperfusión por estadios o graduada después de 15 minutos, revertía de manera completa la disfunción post–isquémica sistólica, es decir el aturdimiento del miocardio. Okamoto F et al,42 ya en 1986 habían publicado que cuando la reperfusión se hacía con presiones controladas de 40 a 50 mmHg daba como consecuencia una restauración parcial de la función segmentaria del miocardio, reducción del edema y un 28.% de abatimiento del tamaño del infarto. En aquellos estudios experimentales que denotaron beneficio al haberse controlado la reperfusión, ésta se realizó inicialmente a presiones y a flujos bajos, por el contrario en tales experiencias cuyos resultados se consignaron como negativos, el reflujo coronario o fue incrementado por períodos cortos o fueron fijados a niveles que si bien impedían la hiperemia eran situados a valores normales de presión de reperfusión coronaria.54,55 En el estudio de Vintén – Johansen J et al,45 donde se instituyó la reperfusión a bajo nivel de flujo y de presión coronaria y se hizo un incremento paulatino de estos parámetros en un período de 30 minutos, dio como resultado que se redujo el tamaño del infarto en un 67.% y se atenuó su extensión transmural al ser comparados con aquellos corazones en los que la reperfusión se concretó de manera abrupta. Además se preservó la función segmentaria en la condición post–isquémica en 30.% y se previno en esta misma etapa la disfunción diastólica. Estos y otros datos sugieren, que el modo hidromecánico en el que se haga la reperfusión es importante, tanto en determinar la función post–isquémica y la magnitud final del infarto. Es relevante señalar que Vintén – Johansen J et al,45 encontraron en el grupo de animales que recibió la reperfusión de manera abrupta, en el segmento del tejido infartado que el flujo miocárdico post–isquémico del subendocardio, ésta se restauró de una manera incompleta. Este aspecto fisiopatológico ya había sido reportado por Becker LC et al,56 donde el fenómeno de no – reflujo o más apropiadamente designado como zona de bajo – flujo, se consideró como un proceso de deterioro post–reperfusión y que extiende el tamaño del infarto más allá del que ya se había pre–establecido en el tiempo de la isquemia previa. De tal manera que, el "core" del infarto producto del período de la isquemia se ve agrandado en los momentos iniciales de la reperfusión, dado por mecanismos que provocan daño microvascular, lo que declina el aporte sanguíneo con la consecuente necrosis de un área que se encontraba con aparente viabilidad. Tal declinación del flujo coronario en el tejido infartado acontece tanto en el sub como en el epicardio del tejido abruptamente reperfundido. Por otro lado, se sabe que hay un período de hiperemia al efectuar la reperfusión no graduada del tejido infartado, fase que precede a la declinación del flujo coronario en la región del sub–epicardio, con la consiguiente extensión del infarto (lo que indica un daño por reperfusión en parte hidrodinámico). Situación que es prevenida en la cohorte en el cual ésta se ha efectuado de una manera gentil y en donde se ha visto que la fase de hiperemia se produce o aparece entre los 60 y los 120 minutos de haberse efectuado la reperfusión, lo que a la vez contrasta con la caída de los flujos que se observan en el tejido miocárdico que se reperfunde de manera abrupta. La preservación de la perfusión tisular y la reducción del tamaño del infarto, cuando la reperfusión se hace de manera controlada se puede llevar a cabo, por varios mecanismos que hacen que se conserve la permeabilidad microvascular. Al haberse limitado la presión de perfusión y el flujo coronario, seguramente se produce una alteración transitoria de las fuerzas de Starling que se ejercen para la migración de los líquidos a través de la barrera endotelial, la que está debilitada por la isquemia precedente, situación hidrodinámica que atenúa la salida de los líquidos del compartimiento vascular hacia la del área intersticial. Estado que a su vez reduce la compresión perivascular de la micro–circulación, lo que en consecue ncia mejoraría la atenuada y comprometida perfusión microvascular. Autores como Vinten–Johansen J et al,45 han demostrado que el controlar la reperfusión en su etapa temprana evita la contractura segmentaria anormal observada en los diseños experimentales, a diferencia de cuando ésta se hace de manera abrupta, la que de estar presente impediría el curso normal del flujo coronario. También, se han citado otros mecanismos cito–protectores que pueden ser los responsables de haber reducido la magnitud de la necrosis en los contextos donde se ha procedido a controlar la presión y el flujo de la reperfusión. Así la restauración gradual del flujo coronario, puede haber retardado el lavado de la adenosina endógena, situación que a concentraciones mayores de ésta, favorece su tiempo de exposición a las células endoteliales, a los miocitos y a los leucocitos. Toombs CF et al,57 han consignado que la adenosina inhibe la activación de los neutrófilos, su adherencia al endotelio vascular y abate el tamaño del infarto. También el incremento gradual del flujo coronario debe a la vez, prolongar el tiempo de la acidosis tisular, lo que crea un ambiente de restauración progresiva del pH del tejido. En el informe de Hori M et al,54 se ha demostrado que la reperfusión con sangre acida, lo que mimetiza la reversión gradual del pH, mejora el acortamiento fraccional del miocardio al ser comparado con los grupos en los que se hace una atenuación muy rápida del medio ácido. Otra causa que se ha invocado para explicar la reducción de la necrosis, son los efectos cardíacos protectores contra la toxicidad celular que brinda el factor relajante derivado del endotelio (EDRF) u óxido nítrico. Así la reperfusión paulatina y graduada, puede evitar la disfunción celular endotelial temprana al preservar la elaboración del EDRF, lo que provee de protección a los miocitos del daño post–reperfusión. Aunque varios estudios experimentales han establecido que la necrosis se hace aparente a los 20 – 30 minutos de la oclusión de la arteria coronaria y se sabe que la disquinesia persiste por días a semanas después de períodos cortos de oclusión reversible. Los estudios de Vintén – Johansen J et al,45 han señalado que la reperfusión gentil de los segmentos hace que éstos sean capaces de generar suficiente trabajo cuando se comparan con las áreas del miocardio que por el contrario la recibieron de forma abrupta, en donde la función se deteriora de manera muy significativa (Fig. 5). Se ha determinado que en los animales de experimentación reperfundidos de una forma abrupta, los segmentos del miocardio tienen reducida las dimensiones diastólicas finales, menor distensibilidad diastólica y posiblemente una mayor limitación para recurrir al mecanismo de Frank–Starling. A manera de contraste, en la otra variedad de reperfusión, los fragmentos resultan más complacientes, tienen un diámetro diastólico final mayor y en consecuencia el trabajo segmentario puede ser desarrollado de una manera superior por estos grupos musculares. Glower DD et al,58han expuesto que la magnitud del desarrollo del trabajo post–isquemia segmentaria depende en parte de si, se han conservado las propiedades de la distensibilidad diastólica. Los mecanismos que preservan la distensibilidad en los animales donde se ha controlado la reperfusión parecen incluir la atenuación del edema y el que se impida la sobrecarga del calcio, cuyas condiciones acontecen de manera significativa anormal en la reperfusión no controlada, siendo causas ambas de la disminución de la distensibilidad diastólica del miocardio.
El papel del poro de transición en la permeabilidad mitocondrial (PTPM)
Éstos se han implicado en los últimos años en la génesis del fenómeno del daño post–reperfusión, estructuras que se encuentran ubicados en la membrana interna de la mitocondria. Se consideran poros no específicos, mismos que de acuerdo a las investigaciones de Kroemer G et al,59 al abrirse producen por el mecanismo de apoptosis/necrosis el daño final del elemento celular en cuestión. Al abrirse permiten la entrada de agua y de solutos, lo que acrecienta el tamaño y el volumen de la matriz, con ruptura de la membrana externa mitocondrial, deja libre al Citocromo C el que a su vez inicia el proceso de la apoptosis. La apertura de los PTPM desacopla a la mitocondria dando como consecuencia hidrólisis del ATP con el consecuente colapso del potencial de acción de la membrana. El PTPM se ha demostrado que permanece cerrado durante los episodios de isquemia y sólo se abre en los primeros minutos de la reperfusión. Esto acontece cuando las condiciones para su apertura lo favorecen como son las altas concentraciones de la carga de calcio y del fósforo mitocondrial, del ATP depletado, si hay estrés oxidativo y se ha acrecentado o corregido el pH de la matriz.60 La participación de los PTPM como elementos mediadores del daño post–reperfusión, se ha visto apoyada por el hecho de que la utilización de la Ciclosporina A inhibe su apertura en la fase de la reperfusión, lo que ha resultado finalmente en una situación cardio–protectora.59–65 Se ha aclarado que la inhabilitación de estos poros privan la liberación del Citocromo C y en consecuencia el desarrollo de la apoptosis. Hausenloy DJ et al,65 han manifestado que la inhibición de los PTPM en los primeros minutos de la reperfusión, utilizando sanglifehrin –A (que es un potente inmuno–supresor superior a la Ciclosporina A), limita el tamaño del infarto y protege a los miocitos del estés oxidativo. También se ha descubierto que el uso del sanglifehrin –A, mejora la recuperación de la función ventricular y reduce la liberación de la LDH. Con fundamento a las siguientes observaciones en las que: 1. Zhao ZQ et al,12 han indicado que el fenómeno de pre–acondicionamiento y el de post–acondicionamiento, han resultado de manera similar cardio–protectores y que la magnitud del infarto después de la isquemia – reperfusión es debido al daño post–reperfusión; 2. que Griffiths EJ y Halestrap AP,6O,61 han expuesto que los PTPM permanecen cerrados durante el proceso de la isquemia pero se abren en la reperfusión temprana y 3. los informes más recientes han consignado que los PTPM están involucrados en el pre–acondicionamiento del miocardio y que el bloqueo de éstos en el fenómeno de reflujo con Ciclosporina –A es cardio–protector;59–63 Argaud L et al,64 investigaron si la apertura de los PTPM pueden jugar un papel relevante en el proceso del post–acondicionamiento. Estos últimos investigadores, demostraron en un modelo experimental de corazón de conejo in vivo, que el postacondicionamiento reduce de manera significativa el tamaño del infarto y que esta protección del miocardio se ve preservada hasta 72 horas post–reperfusión, lo que indica que no sólo simplemente retrasa el daño post– reperfusión, sino que además la limita. Empleando NIM811, que es un inhibidor específico de los PTPM, notaron que la magnitud de protección miocárdica fue similar a la documentada con el proceso mecánico del postacondicionamiento del miocardio. Estos observadores, manifestaron que el proceso de postacondicionamiento retrasa la apertura de los PTPM inducida por el calcio, patrón funcional que es muy similar en su acción al que se ha notado en el fenómeno de pre–acondicionamiento y que mimetiza al documentado en los corazones de conejo aplicado en el momento de la reperfusión cuando se ha empleado NIM811. De acuerdo a las observaciones de Argaud L et al,64 se puede establecer la hipótesis de que el mecanismo del post–acondicionamiento del miocardio, puede alterar la producción de los ROS por la cadena respiratoria y retrasar la apertura de los PTPM. Aunque hay que demostrar de manera cabal, la asociación entre el post–acondicionamiento y la inhibición de los PTPM, los estudios experimentales realizados hasta ahora sugieren que la inhibición PTPM resulta en un muy importante mediador estructural de la cardio–protección encontrada en el mecanismo del post–acondicionamiento.59–65
La activación de la vía del fosfatidilinositol 3–cinasa ( PI3K ) – Akt
Tsang A et al,66 recientemente han aclarado que en el post–acondicionamiento se activa la vía del fosfatidilinositol 3–cinasa (PI3K) – Akt y su blanco final la sintasa inducible del óxido nítrico endotelial y p70S6K. Yang XM et al,67 también han manifestado que la inhibición de la sintasa inducible del óxido nítrico endotelial y el bloqueo de los canales mitocondriales del K –ATP no limitan el tamaño del infarto mediante la aplicación de la secuencia del post–acondicionamiento. Dentro de las nuevas direcciones terapéuticas que han protegido al corazón del daño que ocasiona el binomio isquemia – reperfusión, éstas se han dirigido a salvar la vía enzimática que se ha designado como RISK (Reperfusion Injury Salvage Kinase – pathway).68Esta ruta bioquímica comprende tanto la del fosfatidilinositol 3–cinasa (PI3K) – Akt y la proteína mitógena activada cinasa p44/p42 de naturaleza extracelular. En el año de 2004 Tsang A et al,66 establecieron la hipótesis que el post–acondicionamiento protege al corazón al activar esta ruta RISK y de manera específica el PI3K – Akt, en los primeros minutos de la reperfusión. Situación antes mencionada que logró ser demostrada en el corazón aislado de ratas por este grupo de investigadores. La protección que da esta vía al corazón reperfundido, al valerse del camino RISK,68 resulta similar a la que se ha descrito para la insulina, para la bradiquinina, para la atorvastatina y para el urocortin cuando son administradas en los primeros minutos de la reperfusión. Por lo que estos agentes se pueden considerar que actúan a través de una vía bioquímica (post–acondicionamiento farmacológico). El postacondicionamiento, descrito originalmente en el modelo canino in vivo, también se ha documentado en el conejo, en el corazón de la rata y en los cardiomiocitos aislados de esta última especie. En el primer ensayo donde se investigó este fenómeno, implicó la reducción en el amontono de los neutrófilos activados y una mejoría de la función endotelial como los posibles mecanismos para alcanzarse la protección del miocardio. Sun HY et al,69 han aclarado que en medio de cultivo de cardiomiocitos de neonatos, el post–acondicionamiento reduce la generación de las especies reactivas del oxígeno, la subsiguiente peroxidación de los lípidos y atenúa las concentraciones de calcio tanto intracelular como en el seno de las mitocondrias. Estudios subsiguientes, han demostrado que la protección del miocardio se llega ha alcanzar "en ausencia de los constituyentes de la sangre", lo que sugiere fuertemente que el post–acondicionamiento puede tener de por sí la capacidad de atenuar algunos posibles sucesos en los primeros minutos de la reperfusión. Es decir cabe la posibilidad de haber un efecto directo de éste sobre los miocitos. Kin H et al70 han aclarado utilizando corazones de ratas, que el post–acondicionamiento atenúa la producción de las especies reactivas del oxígeno (ROS) inmediatamente en la reperfusión y que la protección post–acondicionamiento se perdía si ésta se había instituido después de haber transcurrido tan sólo "un minuto" de provocarse la reperfusión. El mismo grupo de investigadores, han demostrado que el post–acondicionamiento hipóxico de los miocitos de ratas neonatas, resulta en una menor cuantía de muerte celular, se atenúa la producción del ROS y se reduce la carga mitocondrial del calcio.71 Datos recientes revelan que la presencia de las proteínas mitógenas activadas cinasa – cinasa (MEK) 1/2 inhibidor PD98059 o el inhibidor del óxido nítrico L–NAME durante el post–acondicionamiento en corazones de conejo in vivo, son capaces de abrogar la protección. Lo que sugiere que las cascadas MEK 1/2 – ERK 1/2 y el óxido nítrico se requieren o se necesitan para poderse alcanzar la tan deseada salvaguarda.72 Sin embargo, los estudios de Yang X et al,72 no han aclarado que el post–acondicionamiento de una manera directa activen ni ERK 1/2, ni la sintasa endotelial del óxido nítrico (eNOS). Como se ha señalado en el inciso correspondiente, la apertura de los poros de transición de la permeabilidad mitocondrial (PTPM), en los primeros minutos de la reperfusión, se ha demostrado que median los procesos de muerte celular y que la inhibición de esta causa resulta cardioprotectora. Situación que ha llevado a postular que el postacondicionamiento protege al corazón al inhibirse la apertura del los PTPM mediante la activación de "Akt y de la eNOS".73,74 E1 postacondicionamiento se puede considerar como un proceso que modifica el daño por reperfusión haciendo a la célula miocárdica y a la mitocondria menos sensibles a las perturbaciones bioquímicas y a las metabólicas que acontecen en el período de transición entre la fase de isquemia y la de la reperfusión. Mas es de hacerse notar que investigadores como Tsang A et al,66 también han indicado que el post–acondicionamiento ha resultado una causa activa al echar a andar las vías de las cinasas como el PI3K–Atk en concordancia con el camino bioquímico RISK.68 Existen diversas fuentes experimentales que apoyan que el post–acondicionamiento, en su modalidad pasiva o mediante mecanismos activos, aplicado en los primeros minutos de la reperfusión pueden dar verdaderas oportunidades de protección al miocardio, lo que puede ser aplicado en el escenario clínico al momento de efectuar la fibrinólisis, los PCI o en la cirugía cardíaca (por ejemplo, en CRVC y en el trasplante del corazón).44,46,59,66,72–78
Su aplicación en el hombre con SICA C E SST
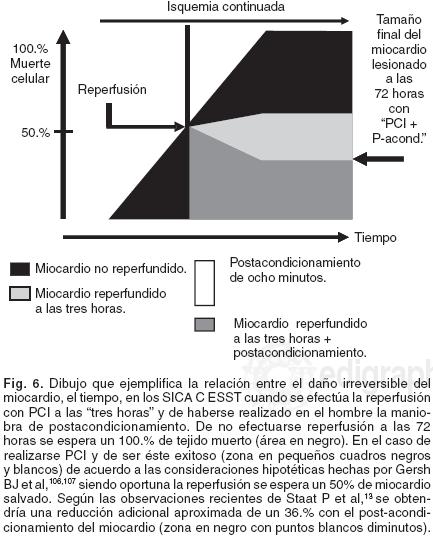
Las aportaciones recientes en el post–acondicionamiento han hecho resurgir los aspectos relacionados con el daño post–reperfusión. Como señalan García–Dorado D y Piper HM,75 en su editorial del año 2006, el deterioro post–reperfusión ha pasado de un estado académico de hibernación a ser una posible ruta terapéutica aplicable al hombre, gracias a los conceptos adquiridos en esta época moderna del análisis del mecanismo del post–acondicionamiento. Así, las oclusiones intermitentes muy breves de la ARI, aplicadas de manera inmediata a la reperfusión han dado como consecuencia el hecho trascendente de limitar el tamaño del infarto, resultando ésta similar en su magnitud a la que se ha obtenido con el pre–acondicionamiento (Figs. 3 y 4). Aspectos del fenómeno de post–acondicionamiento, que están relacionados con la instalación gradual del flujo coronario y que contrasta con la maniobra abrupta de efectuarlo, mas que tienen un protocolo similar para crearlos. Es de consignarse que uno es empleado antes de que se establezca la isquemia y el otro después de que ésta se ha concretado de manera prolongada. Con énfasis se señalará que el momento del pre–acondicionamiento es mucho menos crítico en la aplicación de su diseño, que el que se requiere para que aparezca el de post–acondicionamiento, el cual necesariamente se debe de ejecutar "en los primeros minutos" para que el mecanismo sea efectivo (Fig. 2). La vigencia de este último en el momento de la reperfusión para limitar el tamaño del daño, ha resultado también una demostración que avala la existencia del daño post–reperfusión. La mayoría de los acercamientos farmacológicos que han tendido a limitar el insulto post–reperfusión, han empleado medicamentos que no han sido fáciles de valorar en su efecto cabal, más tratándose del mecanismo de post–acondicionamiento, éste tiene implicaciones prácticas para ser ejecutado en los enfermos que están evolucionando con SICA C ESST y en especial en los que se someten a PCI de manera oportuna con miras a obtener una reperfusión exitosa de la ARI. Con el conocimiento establecido de la protección que brinda el post–acondicionamiento, en el escenario del miocardio con isquemia prolongada, éste en sus efectos es similar en potencia al que ofrece el pre–acondicionamiento y es independientemente de la especie estudiada y también se sabe que el hacerlo no acrecienta la eficacia del post–acondicionamiento.79 Con estos fundamentos Staat P et al,13 lo investigan por vez primera en el año del 2005 en el escenario cardiológico, en treinta enfermos con I AM sometidos a la angioplastíapercutánea transluminal primaria. Los afectados fueron separados de una manera aleatoria a la situación de control (n = 15) y los otros restantes se sometieron al postacondicionamiento (n = 15). Después de realizada la reperfusión mediante la inserción del stent directo (sin dilatación previa) en la ARI, al conjunto control no se le efectuó otro procedimiento y al grupo investigado a continuación sí la maniobra terapéutica de post–acondicionamiento, la que se concretó al minuto de la reperfusión en cuatro episodios caracterizados por un minuto de insuflación del balón de angioplastía y uno de colapso del mismo (Fig. 2). Los investigadores determinaron el área del infarto con base a la cantidad liberada de la creatina–cinasa (CK) en 72 horas. El área bajo la curva de la CK fue reducida de manera significativa en aquellos enfermos en los que se efectuó la maniobra de post–acondicionamiento, al ser comparada con la del grupo control (208,984 ± 25,576 versus 326,095 ± 48,779, unidades arbitrarias, p < 0.05), lo que representó una reducción del 36.% del tamaño del infarto. El grado de la mancha del miocardio usado como marcador de la reperfusión microcirculatorio (TIMI miocárdico), se incrementó con el procedimiento de post–acondicionamiento al ser confrontado con el que se obtuvo en la cohorte control (2.44 ±0.17 versus 1.95 ± 0.27, respectivamente, p < 0.05).13 Este estudio ha demostrado que, la lesión letal post–reperfusión es una entidad clínica relevante y que además sus hallazgos dan soporte a la hipótesis de que es factible prevenirla hasta cierto punto, el daño miocárdico post–reperfusión, lo que eventualmente puede limitar el tamaño del infarto, preservar la función ventricular y tal vez establecer un mejor pronóstico para la vida (Fig. 6). No hay duda que el haberse enriquecido con el conocimiento acerca de estos aspectos fisiopatológicos del deterioro post–reperfusión en la clínica, pasa de una situación patológica que deja de ser una curiosidad del laboratorio, para convertirse en una entidad práctica a considerarse en el escenario de los SICA C ESST. Este estudio es un verdadero parteaguas y ha resuelto parcialmente el cuestionamiento, que se formulara hace 20 años Buckberg D,76 ¿cuándo es que en realidad el músculo cardíaco muere?, demostrándose ahora cuanto contribuye el insulto post–reperfusión en su situación final. Sobresaliente es el concepto que hoy día se puede tener que el mecanismo de post–acondicionamiento, revela el aspecto de un armamentario "endógeno" de protección del daño post–reperfusión, mismo que había sido pasado por alto en el curso de muchos años, lo que a la vez ha generado un interés que se puede considerar explosivo en reanalizar los mecanismos que intervienen en la reperfusión y en buscar nuevas oportunidades terapéuticas en este escenario de los SICA C ESST.


De acuerdo a la opinión de autoridades en el tema como las de Vinten–Johansen J et al,43 hay una serie de cuestionamientos que aparecen en relación al posible daño que se pueda ocasionar en la ARI como producto de las insuflaciones repetidas del balón de angioplastía, como que se produzcan desalojos del material ateromatoso–trombótico o que se ocasionen disecciones del vaso. En el estudio de Staat P et al,13 que es pionero en demostrar el concepto del post–acondicionamiento en los seres humanos, en una población selecta de ellos, la maniobra fue segura al menos de una manera aguda y sin la presentación de episodios adversos después de la inserción del stent.
La técnica empleada en la clínica para alcanzar el estado de post–acondicionamiento por medio del PCI utilizada por Staat P et al,13 fue la siguiente: después de la colocación directa del stent, el balón de angioplastía fue ubicado de manera proximal con el fin de evitar posibles daños y para prevenir microembolias provenientes del trombo como consecuencia de las futuras insuflaciones del balón de angioplastía, mismo que fue ensanchado en cuatro ocasiones por un minuto cada una, a una presión de cuatro a seis atmósferas y alternadas con sesenta segundos de reflujo coronario (Fig. 2). Maniobra que fue seleccionada hasta cierto punto de manera arbitraria, aunque la misma secuencia de oclusión y de reperfusión, fue la que desencadenó el fenómeno de post–acondicionamiento en el corazón del conejo en el estudio de Argaud L et al.64 En el caso de haberse realizado esta maniobra, los investigadores clínicos tuvieron especial cuidado de no obstruir algún ramo lateral de la arteria descendente anterior o de la coronaria derecha. En este sentido de la manipulación del tiempo de la ARI, es menester señalar que el mecanismo de post–acondicionamiento se ha alcanzado en otras especies como en el perro con obstrucciones de 30 y de 10 segundos en las ratas. Lapsos estos últimos que, en los modelos experimentales son capaces de desencadenar las vías protectoras bioquímicas que se han discutido con antelación, mas se desconoce si de manera similar se activan al minuto de haberse hecho la colocación del stent en el hombre y se logren obtener los beneficios en la mitocondria y en especial en los poros de permeabilidad de esta estructura. Lo que si parece seguro es que, tiempos alternados de "cinco minutos de oclusión – reperfusión" fallan en reproducir el fenómeno del post–acondicionamiento del miocardio y precisamente este tipo de esquema fue el que llevó a mantenerlo en el letargo científico hace ya más de 10 años.
Aunque el protocolo utilizado por Staat P et al,13 rindió frutos muy favorables se requiere ahondar en estos aspectos técnicos de la investigación clínica y en otros. Por ejemplo, en relación a los antecedentes ninguno de los enfermos tenía infarto previo y sólo el 20.% de ellos eran diabéticos. Eran sujetos de menos de 62 años, con fracciones de expulsión del ventrículo izquierdo normales (grupo tratado: 52. ± 2%). Por lo que queda la interrogante por indagar, de cuál será el impacto del post–acondicionamiento en aquellos con fracción de expulsión deprimida o con el síndrome de estado de choque o en que proporción de los enfermos con SICA C ESST que van a PCI es posible aplicarlo. También otro aspecto por puntualizar, será si el post–acondicionamiento tiene algún papel en presencia de circulación colateral > 1–2 de Rentrop77o beneficio de efectuarse si el flujo epicárdico se documenta en grados TIMI epicárdico > 1 pero menores de 3. Lo que en principio no luce probable, no obstante hay que recordar que debe de existir un cierto daño miocárdico también reversible, ya que el insulto de la isquemia prolongada estuvo presente, aunque con un área del miocardio en riesgo menor. Galagudza M e investigadores,78 en el corazón aislado de las ratas ha consignado que el mecanismo de post–acondicionamiento puede convertir la fibrilación ventricular a ritmo cardíaco normal. Así mismo Halkos ME et al,79 han notado una menor incidencia de arritmias malignas del tipo de la fibrilación ventricular post–reperfusión en el perro post–acondicionado. Por lo que estos aspectos antiarrítmicos potenciales, también deben de ser considerados en el terreno de la clínica y que al momento se desconocen, lo que tendría particular interés en algunos grupos de enfermos de alto riesgo con SICA C ESST.
La valoración del impacto del postacondicionamiento en el escenario clínico
El dato de mayor valía consignado en el informe de Staat P et al,13 radica en haber demostrado que el tamaño del IAM se abate aproximadamente 36.%. Reducción que fue valorada por determinación enzimática, cifra porcentual que es muy similar a la que investigadores como Kloner RA y asociados,80 y por Ottani F et al,81 han documentado en sus estudios de "pre–acondicionamiento" en el corazón humano. La liberación de la creatina–cinasa (CK) es un método enzimático que se ha validado con respecto a la información que nos brinda la imagenología del SPECT, lo que se ha demostrado en varios estudios y se considera una técnica útil y fácil para poder apreciar el daño miocárdico irreversible en la clínica. Análisis de las curvas que tendrá valor si los tiempos de isquemia, la circulación colateral y el área en riesgo son similares entre los grupos que se consideran como controles y los que son sometidos al procedimiento de post–acondicionamiento. Aunque hay que aceptar que el SPECT usando tecnecio 99 – sestamibi, hasta hoy parece ser el método más adecuado para determinar el área en riesgo de los enfermos con SICA C ESST y que van a ser sometidos a reperfusión de la ARI. Aunque, el área en compromiso en cada sufrido también puede ser estimada al medir la extensión circunferencial de los segmentos contráctiles (ACS) sí se aplica el método de Feild BJ et al.82 Medida que a la vez ha correlacionado de manera satisfactoria con la estimación que se ha hecho con la del SPECT usando tecnecio 99 – sestamibi, como ha sido demostrado por Rogers WJ et al,83 y por Lapeyre AC y colaboradores.84 No obstante es de consignarse que, se ha observado una correlación significativa entre la liberación de la CK y el ACS, la que es muy similar a la que se ha señalado en las preparaciones experimentales donde se usan técnicas de referencia como las tinciones con azul y la de trifeniltetrazolio las que permiten delinear el área en riesgo y la del tejido necrótico.85,86Estas observaciones permiten señalar que el área de ACS se puede emplear en la clínica para valorar el área en riesgo. Con lo antes dicho es de hacerse notar que, la utilización de la CK en el escenario de los SICA C ESST va a tomar nueva y más fuerza en su aplicación práctica en nuestros días. Así los datos que se deriven en estas primeras horas de su determinación, con miras a señalar si en efecto hubo una reducción del tamaño del infarto, deberán ser confirmadas con otras técnicas en las semanas o en los meses venideros con el SPECT o con la resonancia magnética nuclear, con miras a confirmar si en realidad hay una reducción real y sobre todo permanente del tamaño del infarto que se ha sometido al post–acondicionamiento.
Existen otros recursos en la clínica para valorar el impacto del post–acondicionamiento, estos son la graduación de la mancha del miocardio (Blush) y posiblemente la vigilancia oportuna de la evolución del segmento ST particularmente cuando ésta se hace en los primeros 60 a 90 minutos post–reperfusión. En relación al primero, van't Hof AWJ et al,87 y Poli A y colaboradores,88 han hecho que ésta se establezca como un marcador de la perfusión del miocardio en los primeros minutos de haberse hecho la reperfusión de la ARI, al igual ha resultado un indicador del pronóstico a largo plazo para los enfermos con SICA C ESST. Método angiográñco coronario que fue utilizado por Staat P et al,13 para valorar el impacto del postacondicionamiento, encontrando un incremento significativo del mismo en el grupo sometido a la manipulación protectora al ser comparado con la cohorte control (2.44 ±0.18 versus 1.79 ± 0.28, respectivamente , p = 0.02). Lo que traduce una mejoría de la perfusión microvascular post–acondicionamiento del miocardio. En relación al análisis de las modificaciones descendentes del segmento ST, habrá que tomar en consideración el hecho conocido que se ha derivado de los estudios experimentales que el flujo miocárdico puede tener variaciones muy significativas en las primeras 48 horas después de realizada la reperfusión en el seno del área en riesgo, después de un tiempo prolongado de isquemia y en donde se ha efectuado la reperfusión, como lo ha demostrado experimentalmente Rochitte CE et al.89 De acuerdo a las anotaciones hechas, todo indica que afortunadamente sí contamos con métodos clínicos prácticos para poder valorar el impacto de haber utilizado o no, en cohortes similares de enfermos con SICA C ESST, el recurso terapéutico del postacondicionamiento y así poder estimar con certeza los resultados obtenidos de las investigaciones clínicas en este terreno. Aunque la reducción de la amplitud del tamaño del infarto en el hombre parece estar en los linderos bajos de lo que se ha documentado en los modelos animales (25.% al 70.%), esto puede ser producto en parte, de las diferencias en las especies analizadas, que en estas últimas los géneros estudiados están libres de factores comórbidos (diabetes, hipertensión arterial sistémica, hipercolesterolemia, etcétera) y a que los tiempos de isquemia prolongada han sido mucho menores en el escenario de la experimentación animal que en el caso de la experiencia en el humano. Mas es de hacerse notar, que la reducción en el área del infarto en el terreno clínico documentada por Staat P et al13 (36.%), ha resultado muy similar a la observada y comunicada por Mahaffey KW y asociados,90 con la aplicación de la adenosina al momento de realizar la reperfusión con fibrinolíticos en los SICA C ESST.
Aspectos terapéuticos "no mecánicos" del post–acondicionamiento y su aplicación futura en la práctica de los SICA C ESST
No hay duda que las primeras observaciones hechas en el individuo que ha sufrido isquemia prolongada y que se ha sometido a la reperfusión y en donde se incluye la instalación mecánica del post–acondicionamiento, han dado resultados favorables y que por lo tanto abren la potencialidad de un recurso terapéutico para ser utilizado en las salas de hemodinámica en enfermos que acudan oportunamente con SICA C ES ST. La instrumentación mecánica coronaria luce a primera instancia simple y segura para las manos expertas y de enorme potencial clínico por su naturaleza cardioprotectora. Sin embargo, también conocemos que lamentablemente la mayoría de los enfermos con SICA C ESST no se podrán beneficiar con este tipo de tratamiento, lo que abarca de la misma manera a los que no son seleccionados para que se les efectúe PCI. Por lo tanto, los conocimientos que existen acerca de los mecanismos fisiopatológicos que aparecen en la etapa de reperfusión nos debe de obligar hacer mirar hacia ellos, ya que también se ha señalado que existen las vías de sobrevivencia de las cinasas y su activación al momento de la reperfusión debe de ser considerada. Por ende, ya que coexisten elementos moleculares que median el post–acondicionamiento y es posible que éstos puedan suministrarse al momento de la reperfusión. Téllez JF et al,91 han demostrado que la administración experimental de perezone ha resultado cardioprotectora suministrada antes de que se realice la reperfusión, lo que puede estar vinculado a la liberación del calcio intramitocondrial, lo que favorece la producción del ATP durante la reperfusión. Chávez EC y colaboradores,92 también han encontrado acciones bienhechoras para el ketorolaco contra el desarrollo de arritmias malignas, acción que puede estar mediada por su efecto quelante de los iones de calcio, previniendo así la sobrecarga de éste en las células del miocardio. Parra E y asociados,93 al emplear octilguanidina, que es un catión hidrofóbico, han notado que éste protege del daño post–reperfusión a las ratas,94 lo que puede ser debido a su acción conocida de inhibir los poros no específicos de las mitocondrias, el cual es uno de los mecanismos que se ha analizado en el daño post–reperfusión. La posible inhibición de la sobrecarga del calcio a nivel mitocondrial media en parte, el deterioro post–reperfusión, lo que promueve una selectividad no específica de la permeabilidad de la membrana interna. Bobadilla I et al,95 al comparar ratas hipotiroideas versus hipertiroideas, conociendo que la membrana de la mitocondria es más resistente al daño inducido por el calcio en el caso de las primeras, demostraron por estudios histológicos que las fibras miocárdicas de las ratas hipotiroideas retuvieron más sus características histológicas normales y notaron ausencia de edema en las mismas. Hallazgo experimental que sugiere que tal vez los estados hipotiroideos confieren menos riesgo que los hipertiroideos en el hombre que sea víctima de la isquemia prolongada y posteriormente se trate de beneficiar de la reperfusión. Por lo tanto, hay certidumbre experimental, que con la aplicación de ciertos medicamentos es factible abatir la lesión post–reperfusión. Hay agentes que han demostrado que activan las vías de las cinasas de sobrevivencia, como son la insulina, el péptido –1 similar al glucagón, la eritropoyetina, la sim–vastatina y la atorvastatina.68 Si éstos "mimetizan el post–acondicionamiento", pueden ser suministrados antes de que se pretenda efectuar la reperfusión ya sea por el camino de la fibrinólisis o inclusive de manera adyuvante a los PCI. Partiendo de los mecanismos conocidos, en el terreno experimental, es permisible vislumbrar posibles estrategias farmacológicas que pudieran dar protección a un número mayor de enfermos con SICA C ESST. Por ejemplo, centrarse en aspectos como parece ser la recuperación retardada de la acidosis intracelular al momento de efectuar la reperfusión, ya que el post–acondicionamiento enlentece tal fase de ésta y da protección tanto a los cardiomiocitos como a otro tipo de células al inhibir la hiper–contractilidad, atenuando la activación de la calpaína,96 la que favorece la fragilidad celular y el desprendimiento de la bomba de sodio del sarcolema e inhibiendo la permeabilidad de los poros mitocondriales de transición. De tal manera que si la acidosis celular prolongada es un mecanismo relevante, la reperfusión acidificada mediante la infusión intracoronaria de substancias con estos grados de pH puede ser una medida adjunta o alternativa a las insuflaciones alternas del balón de angioplastía. Todo parece indicar, ahora más que nunca, que tanto la investigación mecanicista como la farmacológica se requiere en los terrenos de la reperfusión y luce potencialmente relevante para la práctica clínica. Las investigaciones de Tsang A et al,66 han dado luz a los mecanismos que son capaces de proteger al tejido miocárdico reperfundido al activar las vías RISK . Tanto el pre– como el post–acondicionamiento activan estos caminos fundamentales, lo que incluye la del fosfatidil –inositol 3 – cinasa Akt y la señal extracelular cinasa reguladora, la activación de los receptores acoplados G– proteínicos y otros que incluyen la fosforilización de la sintasa endotelial del óxido nítrico y la inhibición de la apoptosis se ven promovidos, lo que finalmente es muy deseable. Para activar estos mecanismos en los terrenos experimentales se requiere de la rápida institución de ciclos de 30 segundos de reperfusión y 30 de obstrucción coronaria en los diseños animales (Fig. 2), por lo que desconocemos si tales vías RISK en realidad sí se llegan a activar al minuto de la colocación directa del stent, aspecto que requiere ser verificado en el hombre.43,66 Al igual que en el caso del pre–acondicionamiento isquémico, las rutas protectoras que también acontecen en el post–acondicionamiento convergen en la mitocondria y en los PTPM, abriéndose estos &uacut e;ltimos en los primeros minutos de la reperfusión como una respuesta a la sobrecarga de calcio, al estrés oxidativo y a la reducción de la adenosina. Tanto el pre– como el post–acondicionamiento protegen al corazón al inhibir la permeabilidad de los PTPM. El efecto bienhechor del post–acondicionamiento está directamente relacionado o adicionalmente vinculado al efecto beneficioso anti–inflamatorio o a la acción anti–oxidante al disminuir los niveles extracelulares de metabolitos nocivos como los protones y los lactatos, pero también es producto de un "lavado lento de la adenosina", la que es un mediador bien conocido del pre–acondicionamiento isquémico.14 Así como se ha transportado la secuencia mecánica del pre–acondicionamiento isquémico al inicio de la reperfusión, de igual manera se ha buscado la posología o la combinación farmacológica que <<mimetice la acción del pre–acondicionamiento>>, no obstante ahora trasladado al escenario inmediato de la reperfusión. Uno de estos esquemas incluye a la adenosina sola o ésta más la lidocaína,97 ya que se ha percibido que protege del daño celular tanto en la isquemia como en la reperfusión experimental, sin embargo no se ha empleado en el hombre con SICA C ESST. El único antecedente que existe de asociar la adenosina en la reperfusión (fibrinólisis en este caso) es el estudio AMISTAD.90 Las investigaciones de Kerensky RA et al,98 realizadas en el hombre acerca del posible papel de la adenosina al momento de efectuar angioplastía se concretaron por vez primera en el año de 1995. Estos autores, demostraron que la administración de un bolo de 100 microgramos de adenosina antes de realizar la primera insuflación del balón de angioplastía falló en abatir la gravedad de la isquemia (juzgada por la desviación del segmento ST y por la intensidad del puntaje del dolor) al ser comparada con la documentada en el grupo control, mas sí ocasionó disminución del segmento ST entre la primera y la segunda insuflación del balón, lo que no se documentó en la cohorte control. Investigaciones posteriores de Leesar MA et al,99 efectuadas en 1997 han demostrado que en enfermos sometidos a la angioplastía por tener lesiones coronarias críticas (sin SICA C ESST) han consignado que la adenosina sí es capaz de pre–acondicionar el miocardio humano, que el pre–tratamiento con ella es efectiva (e inclusive más que el crear el propio pre–acondicionamiento) y que en este escenario en particular, se puede emplear de manera profiláctica para atenuar la isquemia en los sufridos que van a ser sometidos a PCI de la arteria descendente anterior. Assali AR et al,100 en el año del 2000 al emplear adenosina en el escenario del IAM y valorar su impacto en su posible papel de abatir la incidencia del fenómeno del no flujo encontraron que el medicamento era capaz de disminuir su presencia. Por lo tanto, hasta el momento la aplicación de la adenosina en el hombre con SICA C ESST, no se ha hecho en el instante de efectuar el PCI con miras a documentar si en realidad ésta mimetiza el post–acondicionamiento o lo favorece. Aspecto que merece investigarse en el humano con este medicamento, de igual manera con los agentes que abren los canales de K–ATP, con los inhibidores de la bomba de Na+/ K+, como así lo sugieren en su excelente revisión acerca del tema de la protección del miocardio durante el IAM publicada en el año del 2004 por Kloner RA y Rezkalla SH.101 No podemos dejar pasar por alto, las importantes contribuciones que se han hecho en el campo de la anestesiología en los escenarios del pre– y del post–acondicionamiento del miocardio isquémico. Bell SP et al,102han expuesto que la estimulación de los receptores delta opioides protegen al miocardio isquémico de una manera similar como lo hace el pre–acondicionamiento y ésta es vía de los canales mitocondriales de K –ATP. Agentes como el isofluorano, el sevofluorano y "la morfina" se consideran los más prometedores en inducir el pre–acondicionamiento o el postacondicionamiento.103,104 Es más, Weihrauch D et al,105 en el año del 2005 han indicado experimentalmente, que la morfina induce el "post–acondicionamiento" al activar la vía PI3K, por lo que los agentes mencionados son una verdadera esperanza para llegar a "mimetizar" en el futuro al post–acondicionamiento en el escenario de los SICA C ESST. De acuerdo a las observaciones experimentales hechas en el Departamento de Farmacología del Instituto Nacional de Cardiología de México, señalan que es posible que el remifentanil ofrezca un efecto cardioprotector, aspecto que deberá de corroborarse en un futuro próximo cercano. Aspecto que puede complementarse en la investigación del post–acondicionamiento al existir experiencia con el uso del Ru–360 (ruthenium amine complex), que al inhibir los poros no específicos de la membrana mitocondrial previenen del daño irreversible post–isquémico en el animal de experimentación.106–109
¿Cambiará la aplicación del post–acondicionamiento en la clínica la posición de la pendiente de la curva de la extensión del miocardio salvado?
En la época actual de la cardiología moderna para el tratamiento de los enfermos con SICA C ESST, después de una larga controversia, se acepta por la mayoría de los investigadores del tema, que los PCI resultan el mejor abordaje para realizar la reperfusión, particularmente cuando la logística de los centros es apropiada tanto en la prontitud de efectuarlos como cuando el entrenamiento que existe para concretarlos es óptima.110 Sin embargo, para aquellos aquejados que lleguen de manera muy temprana, dentro de las dos primeras horas de haber iniciado sus síntomas, la administración de la terapia fibrinolítica acertada se puede preferir ya que se ha visto asociada a una buena evolución clínica. Esta conducta se favorece en especial si en los centros de atención la aplicación de los PCI se ve retrasada. Tales circunstancias inclinan hacia la fibrinólisis, si la insuflación del balón se alcanza después de los 60 minutos de que se hubiera iniciado la terapia lítica.111,112 Mas de manera universal debemos de recordar que aún en los países desarrollados, los centros de atención que pueden brindar los PCI como recurso terapéutico no es superior al 20.%, que aún contando con estas facilidades la logística de los PCI es por naturaleza compleja e inclusive existe cierta variación en su técnica entre diferentes regiones y centros del mundo.113 Las opciones con las que cuentan los enfermos admitidos a los hospitales comunitarios o de segundo nivel incluyen la administración de fibrinolíticos seguida de una conducta de vigilancia y de espera de la evolución clínica del afectado. Otras circunstancias que existen son PCI sin apoyo quirúrgico en el centro, transferencia a un hospital terciario para efectuar PCI primaria o la aplicación de la llamada estrategia fármaco – invasiva. Ésta incluye la administración de un fibrinolítico y de un inhibidor de la glucoproteína Ilb/IIIa (solos o combinados) como una medida para restablecer de un modo parcial el flujo coronario de la ARI antes de realizar el PCI.114,115 Cuando se toma en consideración: 1. la reducción de la mortalidad esperada y 2. el tiempo transcurrido entre el inicio de los síntomas a la aplicación de la estrategia de reperfusión, autores como Gersh JB, et al116 han construido una gráfica hipotética de esta relación (Fig. 7). En ésta existe un tiempo crítico (dentro de las primeras dos y media a tres horas) en el cual la meta es "salvar miocardio" y pasado este lapso el fin de la reperfusión se convierte en "mantener la ARI abierta", en esta última condición con muy poca probabilidad de amparar al miocardio. La extensión de miocardio preservado será mayor en las primeras tres horas, situación que es descendiente y la posibilidad será menor del 20.% pasado este lapso para considerarse irrelevante después de las 12 horas. La duración crítica del primer período será influenciado por los siguientes factores: la presencia de circulación colateral efectiva, si existió isquemia pre–acondicionada, por las demandas miocárdicas de oxígeno y por la duración de la isquemia "sostenida". En el caso de que la estrategia de reperfusión se retrase, es por lo tanto esperable que se obtenga menos tejido salvado, se acreciente la mortalidad y sólo se obtenga como meta la apertura de la ARI.1,114,115 Si se ha aplicado una estrategia de tipo fármaco –invasiva (PCI facilitado) se presume que la curva se desplazará ahora, hacia la derecha, por lo que se espera beneficio. Efecto deseable que será mayor cuanto más se traslade la curva a la posición antes señalada.114,115 Por otro lado, es sumamente interesante mencionar que los tiempos del inicio del dolor–reperfusión fueron del orden de los 320 + 40 minutos en los enfermos estudiados por Staat P et al,13 es decir de 5.3 horas y en esta cohorte con esa "ventana terapéutica" se obtuvo beneficio con el post–acondicionamiento (reducción del área de la curva enzimática de la CK), lo que en los modelos experimentales no ha rebasado el lapso de isquemia prolongada "a la hora con 30 minutos" (Fig. 2).12,43,66–68,79 Lo que nos hace plantear la hipótesis: si en pacientes con menor tiempo de evolución de isquemia prolongada y sin circulación colateral efectiva, el beneficio del recurso del post–acondicionamiento será también de mayor cuantía o acaso será igual (ventana de post–acondicionamiento) (Fig. 7). Además, mirando hacia el otro extremo clínico es de esperarse que aquellos aquejados que lleguen con retraso (desafortunadamente la inmensa mayoría en la clínica hoy día), para aplicarse la reperfusión y se les practique además el post–acondicionamiento, se acreciente el beneficio del PCI y no sólo se pretenda obtener la gracia que hoy día se aspira de mantener la ARI abierta.113–115 Asimismo, queda por analizar, si con la realización de la maniobra de post–acondicionamiento aplicada a los grupos de los enfermos con SICA C ESST, que se hayan estratificado como de riesgo alto y en particular los que tengan grados muy bajos o nulos de circulación colateral, pueda ésta cambiar su habitual mal curso clínico hospitalario o el post–nosocomial116,117
Nos preguntamos si la aplicación adecuada del post–acondicionamiento llegará a ser una forma terapéutica hasta cierto punto de "compensar", el detrimento que ocasionan los frecuentes retrasos con los que llegan los enfermos con SICA C ESST en la práctica diaria, consideración hipotética que ahora semblanteamos y que habrá que demostrar.
Comentarios finales
Es interesante y al mismo tiempo lamentable señalar que desde hace ocho años Piper HM, García–Dorado D y Ovize M,8 en su trabajo de revisión "A fresh look atreperfusion injury", marcaban con toda claridad que la administración intravenosa o la intracoronaria de recursos cardio–protectores deben de ser valorados justo al final de la primera insuflación del balón de angioplastía y sea hasta ahora que se haya ensayado en el hombre este conocimiento por Staat P et al,13 en los SICA C ESST. Lapso entre las observaciones básicas y la práctica que penosamente fue excesivamente prolongado. Mas ahora, con todo el nuevo y sólido fundamento experimental que ha revivido el letargo que sufrió el mecanismo del post–acondicionamiento, cuya implementation en las salas de hemodinámica luce seguro, nada oneroso, ni consumidor de más de "unos cuantos minutos", mas sí empleados de manera muy precisa, es menester que se adquiera una fuerte tendencia a la investigación clínica y a su uso para asentar más la información que nos ha brindado Staat P et al.13En vista de que en el contexto de la medicina real cardiológica, los PCI no son aplicables a toda la población con SICA C ESST, es menester intensificar la investigación para entender mejor los mecanismos moleculares, buscar la medicación o la combinación de ellas que "mimeticen al mecanismo de post–acondicionamiento", con miras a que se puedan destinar a la generalidad de ellos y finalmente abatir las consecuencias deletéreas del daño letal post–reperfusión. Con todo esto se podría derrumbar la morbi–mortalidad de los SICA C ESST que hoy luce estancada, a pesar de los enormes avances que se han logrado en este complejo escenario de la cardiopatía isquémica aguda. Creemos que no debemos de "dejar caminar el tiempo innecesario" para aplicar las claras enseñanzas que nos ha dado la medicina experimental básica, para así llevarlas a la cabecera del enfermo en la clínica, guardando siempre las premisas de la más estricta bioética, pues parece existir una nueva luz en el túnel de la reperfusión al aplicar el post–acondicionamiento del miocardio en los enfermos con SICA C ESST.
Referencias
1. Lupi HE, González PH, Juárez UH, Chuquiure E, Vieyra G, Martínez SC: La meta de la reperfusión en los síndromes isquémicos coronarios agudos con elevación del segmento ST. <<Lo que hay más allá del flujo TIMI3 epicárdico>>. Arch Cardiol Mex 2002; 72: 311–349. [ Links ]
2. Jennings RB, Reimer KA: Factors involved in salving ischemic myocardium effect of reperfusion of arterial blood. Circulation 1983; I25–I–36. [ Links ]
3. ISIS–2 (Second International Study of Infarct Survival) Collaborative Group. Intravenous streptokinase given 0–4 hours of onset of myocardial infarction reduced mortality in ISIS–2. Lancet 1987; 1:502. [ Links ]
4. McGovern PG, Pankow JS, Shahar E, Doliszny KM, Folsom AR, Blackburn H, Luepker RV: Recent trials in acute coronary disease mortality, morbidity, medical care and riskfactors: The Minnesota Heart Survey Investigators. N Engl J Med 1996; 334: 884–890. [ Links ]
5. Thompson PL, Fletcher EE, Katavatis V: Enzymatic indices of myocardial necrosis: influence on short —and long—term prognosis after myocardial infarction. Circulation 1979; 59: 113–119. [ Links ]
6. Braunwald E: Myocardial reperfusion, limitations of infarct size, reduction of left ventricular dysfunction and improved survival: should the paradigm be expanded? Circulation 1989; 79:441–444. [ Links ]
7. Braunwald E, Kloner RA: Myocardial reperfusion: a double –edge sword? J Clin Invest 1985; 76: 1713–1719. [ Links ]
8. Piper HM, García–Dorado D, Ovize M: A fresh look at reperfusion injury. Cardiovasc Res 1998; 38: 291–300. [ Links ]
9. Kloner RA: Does reperfusion injury exist in humans ? J Am Coll Cardiol 1993; 21: 537–545. [ Links ]
10. Ferrari R, Hearse DJ: Reperfusion injury: does it exist and does it have clinical relevanc ? J Thromb Thrombol 1997; 4: 25–34. [ Links ]
11. Murry CE, Jennings RB, Reimer KA: Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 1124–1136. [ Links ]
12. Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten–Johansen J: Inhibition of myocardial injury by ischemic post–conditioning during reperfusion: comparison with ischemic preconditioning. Am Physiol Heart Circ Physiol 2003; 285: H579–H588. [ Links ]
13. Staat P, Rioufol G, Piot C, Cottin Y, Tri Cung T, L'Huilier I, et al: Postconditioning the human heart. Circulation 2005; 112: 2143–2148. [ Links ]
14. Bolli R: Myocardial stunning in man. Circulation 1992; 86: 1671–1691. [ Links ]
15. Hearse DJ, Humphrey SM, Chain EB: Abrupt reoxygenation of the anoxic potassium –arrested perfused rat heart: a study of myocardial injury. J Moll Cell Cardiol 1983; 15: 67–73. [ Links ]
16. Ganote CE: Contraction band necrosis and irreversible myocardial injury. J Moll Cell Cardiol 1983; 15: 67–73. [ Links ]
17. Ganote CE, Vander Heide RS: Importance of mechanical factors in ischemic and reperfusion injury. En: Piper HM (Ed.) Pathophysiology of severe ischemic myocardial injury. Dordrecht: Kluwer, 1990: 337–355. [ Links ]
18. Siegmund B, Zude R, Piper HM: Recovery of –anoxic reoxygenated cardiomyocytes from severe calcium overload. Am J Physiol 1992; 263: H1262–H1269. [ Links ]
19. Siegmund B, Koop A, Klietz T, Schwartz P, Piper HM: Sarcolemmal integrity and metabolic competence of cardiomyocytes under anoxia –reoxygenation. Am J Physiol 1990; 258: H285–H291. [ Links ]
20. Siegmund B, Klietz T, Schwartz P, Piper HM: Temporary contractile blockade prevents hyper–contracture in anoxic –reoxygenated cardiomyocytes. Am J Physiol 1991; 260: H426–H435. [ Links ]
21. Ladilov YV, Siegmund B, Piper HM: Simulated ischemia increases the susceptibility of rat cardiomyocytes to hypercontracture. Circ Res 1997; 80: 69–75. [ Links ]
22. Gao WD, Liu Y, Mellgren R, Marban E: Intrinsic myofilament alterations underlying the decreased contractility of stunned myocardium. A consequence of Ca2+ –dependent proteolysis? Circ Res 1996; 78: 455–465 . [ Links ]
23. Siegmund B, Schlack W, Piper HM: Halothane protects cardiomyocytes against reoxygenation–induced hypercontracture. Circulation 1997; 96: 4372–4379. [ Links ]
24. Schlack W, Hollmann M, Stunneck J, Thämer V: Effect of halothane on myocardial reoxygenation injury in the isolated rat heart. Br J Anaesth 1996; 76: 860–867. [ Links ]
25. Preckel B, Schlak W, Comfëre T, Borthel H, Thämer V: Effects of inhalation anaesthetics on myocardial reperfusion injury in vivo. Pflügers Arch 1997; 433: R15. [ Links ]
26. Ladilov YV, Siegmund B, Piper HM: Protection of the reoxygenated cardiomyocyte against hypercontracture by inhibition of Na+/H+ exchange. Am J Physiol 1995; 268: H1531–H1539. [ Links ]
27. Bugge E, Ytrehus K: Inhibition of sodium –hydrogen exchange reduces infarct size in the isolated rat heart –a protective additive to ischemic preconditioning . Cardiovasc Res 1995; 29: 269–274. [ Links ]
28. Klein HH, Pich S, Bohle RM, Wollenweber J, Nebendahl K: Myocardial protection by Na+/H+ exchange inhibition in ischemic reperfused porcine hearts. Circulation 1995; 92: 912–917. [ Links ]
29. García–Dorado D, González MA, Barrabés JA, Ruiz–Meana M, Solares J, Lidón RM, et al: Prevention of ischemic rigor occlusion by inhibition of Na+– H+ exchange. Cardiovasc Res 1997; 35: 80–89. [ Links ]
30. Inserte J, García–Dorado D, Ruiz–Meana M, Solares J, Soler–Soler J: The Na+– H+ exchange occurring during hypoxia in the genesis of reoxygenation–induced myocardial oedema. J Moll Cell Cardiol 1997; 29: 1167–1175. [ Links ]
31. García–Dorado D, Oliveras J: Myocardial edema: a preventable cause of reperfusion injury. Cardiovasc Res 1993; 27: 1555—563. [ Links ]
32. Schlüter KD, Jakob G, Ruiz–Meana M, García –Dorado D, Piper HM: Protection of reoxygenated cardiomyocytes against osmotic fragility by NO donors. Am J Physiol 1996; 271: H428–H434. [ Links ]
33. Ruiz–Meana M, García–Dorado D, González MA, Barrabés JA, Soler–Soler J: Effects of osmotic stress on sarcolemmal integrity of isolated cardiomyocytes following transient metabolic inhibition. Cardiovasc Res 1995; 30: 64–69. [ Links ]
34. Kloner RA, Reimer KA, Willerson JT, Jennings RB: Reduction of experimental myocardial infarct size with hyperosmotic mannitol. Proc Soc Exp Biol Med 1976; 151: 677–683. [ Links ]
35. Ganote CE: Cell–to–cell interactions contributing to the oxygen paradox. Bas Res Cardiol 1985; 80: 141–146 . [ Links ]
36. Frank JS, Brandy AJ, Fransworth BS, Mottino G: Ultrastructure and function of isolated myocytes after calcium depletion and repletion. Am J Physiol 1986; 250: H265–H275. [ Links ]
37. Fliss H, Gattinger D: Apoptosis in ischemic and reperfused rat myocardium. Circ Res 1996;79: 949–956. [ Links ]
38. Veinot JP, Gattinger DA, Fliss H: Early apoptosis in human myocardial infarcts. Hum Pathol 1997; 28: 485–492. [ Links ]
39. Gotttlieb RA, Burleson KO, Kloner RA , Ba–bior BM, Engler RL: Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 1994; 94: 1621–1628. [ Links ]
40. Buckberg GD: Myocardial protection: an overview. Semin Thorac Cardiovasc Surg 1993; 5: 98–106. [ Links ]
41. Vinten–Johansen J, Edgerton TA, Howe HR, Gayheart PA, Mills SA, Howard G, Cordell AR: Immediate functional recovery and avoidance of reperfusion injury with surgical revascularization of short–term coronary occlusion. Circulation 1985; 72: 431–439. [ Links ]
42. Okamoto F, Allen BS, Buckberg GD, Buyi H, Leaf J: Reperfusion conditions: importance of ensuring gentle versus sudden reperfusion during relief of coronary occlusion. J Thorac Cardiovasc Surg 1986; 92: 613–620. [ Links ]
43. Vinten–Johansen J, Yellon DM, Opie LH: Post–conditioning. A simple, clinically applicable procedure to improve revascularization in acute myocardial infarction. Circulation 2005; 112: 2085–2088. [ Links ]
44. Sato H, Jordan JE, Zhao ZQ, Sarvotham SS, Vinten–Johansen J: Gradual reperfusion reduces infarct size and endothelial injury but augments neutrophil accumulation. Ann Thorac Surg 1997; 64: 1099–1107. [ Links ]
45. Vinten–Johansen J, Lefer DJ, Nakanishi K, Johnston WE, Brian CA, Cordell RA: Controlled coronary hydrodynamics at the time of reperfusion reduces postischemic injury. Coron Artery Dis l992;3: 1081–1093. [ Links ]
46. Tsao PS, Aoki N, Lefer DJ, Johnson G III, Lefer AM: Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation 1990; 82: 1402–1412. [ Links ]
47. Piper HM, Schafer AC: The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res 2004; 61: 365–371. [ Links ]
48. Zweir JL, Flaherty JT, Weisfeldt ML: Direct measurement of free radicals generated following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA 1987; 84:1404–1407. [ Links ]
49. Schwartz LM: Ischemic Postconditioning during reperfusion fails to protect against lethal myocardial ischemia –reperfusion injury in pigs. Circulation 2004; 110: 111–106. [ Links ]
50. Farber NE, Pieper GM, Gross GJ: Postischemic recovery in the stunned myocardium after reperfusion in the presence or absence of a flow–limiting coronary artery stenosis. Am Heart J 1988; 116:407–420. [ Links ]
51. Granato JE, Watson DD, Flanagan TL, Gascho JA, Beller GA: Myocardial thallium –201 kinetics during coronary occlusion and reperfusion: influence of method ofreflow and timing of thallium–201 administration. Circulation 1986; 73: 150–160. [ Links ]
52. Granato JE, Watson DD, Flanagan TL, Beller GA: Myocardial thallium–201 kinetics and regional flow alterations with 3 hours of coronary occlusion and either rapid reperfusion through a critical stenosis. J Am Coll Cardiol l987; 9: 109–118 [ Links ]
53. Yamazaki S, Fujibayashi Y, Rajagopalan RE, Meerbaum S, Corday E: Effects of staged versus sudden reperfusion after acute coronary occlusion in the dog. J Am Coll Cardiol 1986; 7: 564–572. [ Links ]
54. Hori M, Kitakaze M, Sato H, Takashima S, Iwakura K, Inoue M, et al: Staged reperfusion attenuates myocardial stunning in dogs: role of transient acidosis during early reperfusion. Circulation 1991; 84: 2135–2145. [ Links ]
55. Kuzuya T, Hoshida S, Yamshita N, Fuji H, Oe H, Hori M, Kamada T, Tada M: Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res 1993; 72: 1293–1299. [ Links ]
56. Becker LC, Ambrosio G, Manissi J, Weisman HF: The no–reflow phenomenon: a misnomer? En: Analysis and simulation of the cardiac system ischemia. Ed: Sideman S . Boca Ratón: CRC Press, 1989: 189–309. [ Links ]
57. Toombs CF, McGee DS, Jonhston WE, Vinten–Johansen J: Myocardial protective effects of adenosine: infarct size reduction with pretreatment and continued receptor stimulation during ischemia. Circulation 1992; 86: 986–994. [ Links ]
58. Glower DD, Schaper J, Kabas JS, Hoffmeister HM, Schaper W, Spratt JA, et al: Relation between reversal of diastolic creep and recovery of systolic function after ischemic myocardial injury in conscious dogs. Circ Res 1987; 60: 850–860. [ Links ]
59. Kroemer G, Dallaporta B, Resche–Rigon M: The mitocondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 1998 ; 60 : 619–642. [ Links ]
60. Griffiths EJ, Halestrap AP: Mitochondrial nonspecific pores remain closed during cardiac ischemia, but open upon reperfusion. Biochem J 1995; 307: 93–98. [ Links ]
61. Griffiths EJ, Halestrap AP: Protection by Cyclosporin A of ischemia/reperfusion –induced damage in isolated rat hearts. J Moll Cell Cardiol 1993; 25: 1461–1469. [ Links ]
62. Xu M, Wang Y, Hirai K, Ayub A, Ashraf M: Calcium preconditioning inhibits mitocondrial permeability transition and apoptosis. Am J Physiol Heart Circ Physiol 2001; 280: H899–H908. [ Links ]
63. Arteaga D, Odor A, López RM, Contreras G, Pichardo J, García E, Arnada A, Chavez E: Impairments by cyclosporin A of reperfusion –induced arrhythmias. Life Sci 1992; 51: 1127–1134. [ Links ]
64. Argaud L, Gateau–Roesch O, Raisky O, Loufouat J, Robert D, Ovize M: Postconditioning inhibits mitocondrial permeability transition. Circulation 2005; 111: 194–197. [ Links ]
65. Hausenloy D, Wynne A, Duchen M, Yellon DM: Transient mitochondrial permeability transition pore opening mediates preconditioning–induced protection. Circulation 2004; 109: 1714–1717. [ Links ]
66. Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM: Postconditioning: a form of <<modified reperfusion>> protects the myocardium by activating the phosphatidylinositol 3–kinase Akt pathway. Circ Res 2004; 95: 230–323. [ Links ]
67. Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV: Multiple, brief coronary occlusions during early reperfusion protects rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol 2004; 44: 1103–1110. [ Links ]
68. Hausenloy DJ, Yellon DM: New directions for protecting the heart against ischemia–reperfusion injury: targeting the reperfusion injury salvage kinase [RISK] pathway. Cardiovasc Res 2004; 61:448–460. [ Links ]
69. Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, Vinten–Johansen J, Zhao ZQ: Hypoxic Postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload. Am J Physiol Heart Circ Physiol 2005; 288: 1900–1908. [ Links ]
70. Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten–Johansen J: Postconditioning attenuates myocardial ischemia – reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res 2004; 62: 74–85. [ Links ]
71. Zhao ZQ, Sun HY, Wang NP, Kerendi F, Guyton RA, Vinten–Johansen J: Hypoxic Postconditioning reduces cardiomyocytes loss by inhibiting reactive oxygen species –triggered mitochondrial calcium overload. Circulation 2003: 108(Suppl IV ): 174. [ Links ]
72. Yang X, Downey JM, Cohen MV: Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by activation of ERK and production of nitric oxide. Circulation 2003; 108[ Suppl IV]: 158. [ Links ]
73. Tsuruta F, Masuyama N, Gotoh Y: Thephosphatidylinositol 3–kinase (PI3K)–Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem 2002; 277: 14040–14047. [ Links ]
74. Balakirev MY, Khamtsov VV, Zimmer G: Modulation of the mitochondrial permeability transition by nitric oxide. Eur J Biochem 1997; 246: 710–718. [ Links ]
75. García–Dorado D, Piper HM: Postconditioning: Reperfusion of <<Reperfusion injury>> after hibernation. Cardiovasc Res 2006; 69: 1–3. [ Links ]
76. Buckberg GD: Studies of controlled reperfusion after ischemia: I. When is cardiac muscle damaged irreversibly? J Thorac Cardiovasc Surg 1986; 92: 483–487. [ Links ]
77. Rentrop KP, Cohen M, Blanke H, Phillips RA: Changes in collateral channel filling immediately after controlled coronary occlusion by an angioplasty balloon on human subjects. J Am Coll Cardiol 1985; 5: 587–592. [ Links ]
78. Galagudza M, Kurapeev D, Minasiana S, Valen G, Vaage J: Ischemic postconditioning: brief ischemia during reperfusion converts persistent ventricular fibrillation into regular rhythm. Eur J Cardiovasc Surg 2004; 25: 1006–1010. [ Links ]
79. Halkos ME, Kerendi F, Corvera JS, Wang NP, Kin H, Payne CS, etal: Myocardial protection with postconditioning is not enhanced by ischemic preconditioning. Ann Thorac Surg 2004; 78: 961–969. [ Links ]
80. Kloner RA, Shook T, Przyklenk K, Davis VG, Junio L, Matthews RV, et al for the TIMI 4 Investigators: Previous angina alters in–hospital outcome in TIMI 4: a clinical correlate to preconditioning? . Circulation 1995; 91: 37–45. [ Links ]
81. Ottani F, Galvani M, Ferrini D, Sorbello F, Limonetti P, Pantoli D, Rusticali F: Prodromal angina limits infarct size: a role for ischemic preconditioning. Circulation 1995; 91: 291–297. [ Links ]
82. Feild BJ, Russell RO, Dowling JT, Rackley CE: Regional left ventricular performance in the year following myocardial infarction. Circulation 1972; 46: 679–689. [ Links ]
83. Rogers WJ, McDaniel HG, Smith LR, Mantle JA, Russell RO, Rackley CE: Correlation of angiographic estimates of myocardial infarct size and accumulated release of creatine kinase MB isoenzyme inman. Circulation 1977; 56: 199–205. [ Links ]
84. Lapeyre AC, Gibson WS, Bashore TM, Gibbons RJ: Quantitative regional wall motion analysis with early contrast ventriculography for the assessment of myocardium at risk in acute myocardial infarction. Am Heart J 2003; 145: 1051–1057. [ Links ]
85. Vivaldi MT, Kloner RA, Schoen FJ: Triphenyltetrazolium staining of irreversible ischemic injury following coronary artery occlusion in rats. Am J Pathol 1985; 121: 522–530. [ Links ]
86. Argaud L, Gateau–Roesch O, Chalabreysse L, Gómez L, Loufouat J, Thivolet–Bejui F, et al: Preconditioning delays Ca2+ induced mitochondrial permeability transition. Cardiovasc Res 2004; 61: 115–122. [ Links ]
87. Van't–Hof AWJ, Liem A, Suryapranata H, Hoorntje JC A, De Boer MJ, Zijlstra F, for the Zwolle Myocardial Infarction Study Group: Angiographic assessment of myocardial reperfusion in patients treated with primary angioplasty for acute myocardial infarction: myocardial blush grade. Circulation 1998; 97: 2302–2306. [ Links ]
88. Poli A, Fetiveau R, Vandoni P, Del Rosso G, D'Urbano M, Seveso G, Cafiero F, De Servi S: Integrated analysis of myocardial blush and ST segment elevation recovery after successful primary angioplasty real–time grading of microvascular reperfusion and perfusion of early and late recovery of left ventricular function. Circulation 2002; 106: 313–318. [ Links ]
89. Rochitte CE, Lima JA, Bluemke DA, Reeder SB, Me Veigh ER, Furuta T, et al: Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation 1998; 98: 1006–1014. [ Links ]
90. Mahaffey KW, Puma JA, Barbagelata NA, Di Carli MF, Leesar MA, Browne KF, et al: Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a Multicenter, randomized, placebo–controlled trial: the Acute Myocardial Infarction Study of Adenosine (AMISTAD) trial. J Am Coll Cardiol 1999; 34: 1711–1720. [ Links ]
91. Téllez JF, Carvajal K, Cruz D, Carabez A, Chavez E: Effect ofperezone on arrhythmias and markers of cell injury during reperfusion in the anesthetized rat. Life Sci 1999; 65: 1615–1623. [ Links ]
92. Chávez E, Téllez F, Pichardo J, Milán R, Cuéllar A, Carbajal K, Cruz D: On the protection by ketorolac of reperfusion –induced heart damage. Comp Biochem Physiol 1996; 115C: 95–100. [ Links ]
93. Parra E, Cruz D, García G, Zazueta C, Correa F, García N, Chavez E: Myocardial protective effect of octylguanidine against the damage induced by ischemia reperfusion in rat heart. Moll Cell Biochem 2005; 269: 19–26. [ Links ]
94. Chavez E, Peña A, Zazueta C, Ramírez J, García N, Carrillo R: Inactivation of mitochondrial permeability transition by octylguanidine and octylamine. J Bioenerg Biomembr 2000; 32: 193–198. [ Links ]
95. Bobadilla I, Franco M, Cruz D, Zamora J, Robles SG, Chavez E: Hypothyroidism provides resistance to reperfusion injury following myocardial ischemia. Int J Biochem Cell Biol 2001; 33: 499–506. [ Links ]
96. Inserte J, García–Dorado D, Heranado V, Soler–Soler J: Calapain –mediated impairment of Na+/K+ –ATPase activity during early reperfusion contributes to cell death after myocardial ischemia. Circ Res 2005; 97: 465–473. [ Links ]
97. Canyon SJ, Dobson GP: Pretreatment with an adenosine A1 receptor agonist and lidocaine : A possible alternative to myocardial ischemic preconditioning . J Thorac Cardiovasc Surg 2005; 130:371–377. [ Links ]
98. Kerensky RA, Kutcher MA, Braden GA, Applegate RJ, Solis GA, Little WC: The effects of intracoronary adenosine on preconditioning during coronary angioplasty. Clin Cardiol 1995; 18:91–96. [ Links ]
99. Leesar MA, Stoddard M, Ahmed M, Broadbent J, Bolli R: Preconditioning of human myocardium with adenosine during coronary angioplasty. Circulation 1997; 95: 2500–2507. [ Links ]
100. Assali AR, Sdringola S, Ghani M, Denkats AE, Yepes A, Hanna GP, et al: Intracoronary adenosine administered during percutaneous intervention in acute myocardial infarction and reduction in the incidence of <<no reflow>> phenomenon. Cathet Cardiovasc Intervention 2000; 51: 27–31. [ Links ]
101. Kloner RA, Rezkalla SH: Cardiac protection during acute myocardial infarction: Where do we stand in 2004? J Am Coll Cardiol 2004; 44: 276–286. [ Links ]
102. Bell SP, Sack MN, Patel A, Opie LH, Yellon DM: Delta opioid receptor stimulation mimics ischemic preconditioning in human heart muscle. J Am Coll Cardiol 2000; 36: 2296–2302. [ Links ]
103. Kato R, Föex P: Myocardial protection by anesthetic agents against ischemia–reperfusion injury: an update for anesthesiologist. Can J Anesth 2002; 49:777–791. [ Links ]
104. Xenopoulos NP, Leesar M, Bolli R: Morphine mimics ischemic preconditioning in human myocardium during PTCA. J Am Coll Cardiol 1998; 31: 65A. [ Links ]
105. Weihrauch D, Krolikowski JG, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS: Morphine enhances isoflurane–induced postconditioning against myocardial infarction : the role of phosphatidylinositol–3–kinase and opioid receptors in rabbits. Anesth Analg 2005; 101: 942–949. [ Links ]
106. Zazueta C, Zafra G, Vera G, Sánchez C, Chavez E: Advances in the purification of the mitochondrial Ca2+ uniporter using the labeled inhibitor 103 Ru –360. J Bioenerg Biomembr 1998; 30: 489–498. [ Links ]
107. Zazueta C, Sosa–Torres ME, Correa F, Garza–Ortíz A: Inhibitory properties of ruthenium amine complexes on mitochondrial calcium uptake. J Bioenerg Biomembr 1999: 551–557. [ Links ]
108. García–Rivas GJ, Guerrero–Hernández A, Guerrero–Serna G, Rodríguez–Zavala J, Zazueta C: Inhibition of the mitochondrial calcium uniporter by the oxo–bridged dinuclear ruthenium amine complex [Ru–360] prevents from irreversible injury inpostischemic rat heart. FEBS 2005; 272: 3477–3488. [ Links ]
109. Correa F, Zazueta C: Mitochondrial glycosidic residues contribute to the interaction between ruthenium amine complexes and the calcium uniporter. Mol Cell Biochem 2005; 272: 55–62. [ Links ]
110. Keeley EC, Boura JA, Grines CL: Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomized trials. Lancet 2003; 361: 13–20. [ Links ]
111. Nallamouthu BK, Bates ER: Percutaneous coronary intervention versus fibrinolytic therapy in acute myocardial infarction. Am J Cardiol 2003; 92: 824–826. [ Links ]
112. Giugliano RP, Barunwald E: Selecting the best reperfusion strategy in ST–elevation myocardial infarction: it's all matter of time. Circulation 2003; 108: 12828–2830. [ Links ]
113. RENASICA, Grupo cooperativo: El Registro Nacional de los Síndromes Isquémicos Coronarios Agudos (RENASICA I). Sociedad Mexicana de Cardiología. Arch Cardiol Mex 2002; 72: S45–S64. [ Links ]
114. Gersh BJ, Anderson JL: Thrombolysis and myocardial salvage: results of clinical trials and the animal paradigm –paradoxic or predictable? Circulation 1993; 88: 296–306. [ Links ]
115. Gersh BJ, Stone GW, White HD, Holmes DR: Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction. Is the slope of the curve the shape of the future. JAMA 2005; 293: 979–986. [ Links ]
116. Lupi HE, Lasses LA, Cosío AJ, Chuquiure EV, Martínez SC, Ortíz P, et al: Acute right ventricular infarction : clinical spectrum , results of reperfusion therapy and short–term prognosis. Coron Artery Dis 2002; 13: 57–64. [ Links ]
117. Hochman JS: Cardiogenic shock complicating acute myocardial infarction: Expanding the paradigm. Circulation 2003; 107: 2998–3002. [ Links ]

