










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.76 supl.2 Ciudad de México abr./jun. 2006
Presentación
De la hipótesis de la trombina a la inflamación. ¿Es una realidad?
From thrombin hypothesis to inflammation. It is reality?
Héctor González Pacheco*
* Unidad de Cuidados Coronarios. Instituto Nacional de Cardiología Ignacio Chávez.
Correspondencia:
Dr. Héctor González Pacheco.
Unidad de Cuidados Coronarios. Instituto Nacional de Cardiología, Ignacio Chávez.
(INCICH, Juan Badiano Núm. 1. Col. Sección XVI, Tlalpan 14080. México, D.F).
Resumen
La ruptura de la placa aterosclerótica y la subsecuente trombosis intracoronaria son los mecanismos responsables para el desarrollo de los síndromes coronarios agudos. Se ha considerado que la inflamación juega un papel importante en la inestabilidad de la placa y que la trombogenicidad está dada por la generación de factor tisular (FT) existiendo interrelación entre estos dos sistemas. El FT modula la generación de trombina que no únicamente participa en la hemostasia y trombosis si no también promueve el proceso inflamatorio a través de los receptores activados de proteasas (PARs) provocando un ciclo vicioso entre la inflamación y coagulación.
Palabras clave: Síndrome coronario agudo. Inflamación. Trombina.
Summary
The classical pathophysiologic concept of the acute coronary syndromes is the coronary artery thrombosis as a consequence of rupture or vulnerable atherosclerotic plaques. Actually, it is also been considered that systemic inflammatory phenomenon play a central role in the plaque instability associated to the atherothrombotic activity of the tissue factor (TF). The throm–botic phenomenon is controlled by tissue factor, stimulating the way of the protease's active receptors (PAR) and cause a negative cycle between inflammation and coagulation.
Key words: Acute coronary syndrome. Inflammation. Thrombin.
Los avances importantes en el conocimiento de la fisiopatología de la aterotrombosis (ATT), se han llevado a cabo en las 2 últimas décadas. A la hipótesis del acumulo de lípidos en su fisiopatología, se han integrado varios factores que contribuyen al inicio y a la evolución de la aterotrombosis. La inflamación y la apoptosis de varias líneas celulares participan en forma predominante en el inicio y progresión de la enfermedad ATT y se ha postulado que el factor tisular es el responsable de la trombogenicidad de la placa aterosclerótica.
La inflamación y la coagulación tienen un papel importante en la fisiopatología de las enfermedades vasculares. Hay numerosas evidencias en las que se puntualiza la interrelación de los dos sistemas, en donde la inflamación no sólo activa a la coagulación, sino que la coagulación también tiene importantes efectos inflamatorios. Tanto una exagerada o insuficiente respuesta puede conducir a una situación en la que la coagulación y la trombosis contribuyan a la enfermedad, como sucede en la trombosis intracoronaria sobre una placa de aterosclerosis rota (Fig. 1).

La ruptura de una placa y la subsecuente formación de un trombo es responsable para el inicio de muchos síndromes isquémicos coronarios agudos (SICA). La magnitud de este proceso trombótico es modulado por diferentes elementos que participan tanto a nivel de la placa como en la trombogenicidad de la sangre. Se ha considerado que la composición y el estado de la placa más que el grado de estenosis, es el principal determinante de la enfermedad ATT.1,2 Estas placas llamadas vulnerables o de alto riesgo propensas a romperse o inestabilizarse consistente de grandes "core" lipídico extracelular, un gran infiltrado inflamatorio predominantemente de macrófagos, un reducido número de células de músculo liso y una capa fibrosa delgada. Una vez que la ruptura de la placa ocurre, el core lipídico altamente trombogénico por un elevado contenido de factor tisular (FT), es expuesto al torrente sanguíneo provocando la formación del trombo intracoronario.3–6.
El iniciador principal en la generación de trombina inducida por la inflamación es modulada por el FT, el cual es considerado el principal regulador de la hemostasia y de la trombosis (Fig. 2). El bloqueo de la actividad del FT completamente detiene la activación de la coagulación inducida por la inflamación como se ha demostrado en modelos experimentales de endotoxemia o bacteremia, en donde los anticuerpos que inhiben el sistema de contacto no afecta la formación de trombina.7,8 El FT es una proteína transmembrana que es expresada en varias líneas celulares del organismo.9 El origen del FT puede ser de diferentes fuentes en situaciones de inflamación. En las placas ateroscleróticas, es generado en gran parte por macrófagos y es activado al entrar en contacto con células apoptoicas principalmente de origen endotelial. La mayoría del FT expresado dentro de la placa de ateroma ha sido en áreas de alta densidad de células apoptoicas y de micropartículas,10 éstas tienen actividad procoagulante por su alto contenido de fosfatidilserina, hace suponer la importancia del FT en la formación del trombo y que puede ser un nuevo objetivo en el tratamiento farmacológico (Fig. 3).


La apoptosis dentro del ateroma compromete a todas las línea celulares.11 A pesar de la iniciación de la muerte celular, las células apoptoicas están relacionadas con la inflamación por favorecer el reclutamiento de otras células inflamatorias.12 Mallat y cols.13 han demostrado la asociación entre la apoptosis y la inflamación, coexistiendo los dos procesos en áreas de ruptura de la placa.13 Ellos mismos han encontrado niveles elevados de micropartículas de membrana de células apoptoicas en la placa aterosclerótica14 así mismo mostraron niveles elevados de micropartículas de membrana con potencial procoagulante en la sangre periférica. En resumen el fenómeno apoptoico es fuertemente asociado con la producción de FT activado que al final conduce a la formación de trombina.
La generación de trombina se hace a raíz de la actividad de FT, es conocido que la trombina es crucial en la enfermedad ATT; es la principal proteasa efectora de la cascada de coagulación y se considera que una reacción importante es la conversión de fibrinógeno a fibrina, además de la activación de cofactores esenciales de la coagulación como son el V, VIII y del factor XI, provocando mayor cantidad de factores IXa y Xa, y por otra parte se ha considerado a la trombina como el principal agonista de la activación plaquetaria. También provoca una respuesta al endotelio vascular, como son cambios de forma y permeabilidad, moviliza moléculas de adhesión a la superficie endotelial y estimula la producción de cito quinas y de sustancias autocoides tales como prostaglandinas y factor de activación plaquetaria (Fig. 4). Sin embargo, la trombina como otras proteínas anticoagulantes como es la proteína C activada, puede activar receptores específicos sobre células mononucleares y endoteliales, promoviendo la producción de citoquinas o la propia apoptosis de células inflamatorias.15

El mecanismo más importante por la cual estas proteasas de la coagulación influyen en la inflamación es a través de la unión con los receptores activados de proteasas (PARs), de los cuales se han identificado 4 tipos (PAR 1 al 4), todos son de la familia de receptores acoplados a la proteína G transmembrana16 y se han identificado en células endoteliales, células mononucleares, plaquetas, fibroblastos y células de músculo liso.17 Los PAR 1, 3 y 4 son receptores de trombina, donde el PAR 2, no une a la trombina pero sí al complejo factor tisular – VIIa, factor Xa y tripsina (Fig. 5).
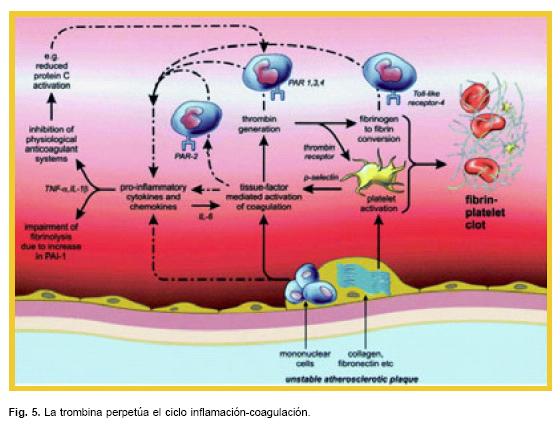
Se conoce la participación de los PARs en la coagulación e inflamación en la trombosis intracoronaria, ratones con deficiencia de PAR–4, muestran in vivo una ausencia de actividad plaquetaria y son protegidos contra trombosis arterial experimental,18 otros efectos que se han demostrado de PAR–1 y PAR–4 es su participación en la hipertrofia de los miocitos y sobre la remodelación cardíaca en la isquemia.19
El conocimiento de los mecanismos moleculares en la enfermedad ATT ha sido trascendente en el desarrollo de terapias antitrombóticas óptimas. El manejo con inhibidores de la trombina como son la heparina no fraccionada (HNF) y actualmente las heparinas de bajo peso molecular (HBPM) han sido la piedra angular en el tratamiento de la enfermedad ATT.
Se ha establecido que la enoxaparina (HBPM) ejerce su efecto antitrombótico principalmente por la inhibición del factor Xa, pero también se ha demostrado que su administración inhibe la generación y la actividad de FVIIa y de la protrombina, independiente de la liberación del inhibidor de vía del factor tisular (TFPI), lo que significa que la enoxaparina disminuye la generación y actividad de trombina.20 Resultados que se correlacionan con el reporte de Keating y cols.21 quienes demuestran que la administración de un inhibidor directo de la trombina (bivalirudina) tiene un efecto más consistente sobre los marcadores de inflamación, disminuyendo su concentración, lo que puede contribuir a una mejor evolución.21
Conclusiones
La trombosis intracoronaria que sucede después de la ruptura de una placa aterosclerótica es el evento más importante para el inicio de un evento coronario agudo. El FT es principal determinante de la trombogenicidad de la placa inestable; así como de la sangre circulante en enfermos ATT, y que este es el factor determinante en la generación de trombina, que no únicamente actúa sobre la cascada de coagulación, sino que participa promoviendo la inflamación, creando un ciclo vicioso entre inflamación y coagulación. Esta interrelación nos ofrece un campo de investigación para nuevas terapias que puedan modificar la excesiva activación de estos dos sistemas.
Referencias
1. Fuster V, Badimon JJ, Badimon L: Clinical–pathological correlations of coronary disease progression and regression. Circulation 1992; 86: III–1–III–11. [ Links ]
2. Badimon JJ, Zaman A, Helft G, et al: Acute coronary syndromes: pathophysiology and preventive priorities. Thromb Haemost 1999; 82: 997–1004. [ Links ]
3. Fuster V, Badimon JJ, Chesebro JH: Atherothrombosis: mechanisms and clinical therapeutic approaches. Vase Med 1998; 3: 231–239. [ Links ]
4. Fuster V, Fay ad ZA, Badimon JJ. Acute coronary syndromes: biology. Lancet 1999; 353 (Suppl 2): SII5–SII9. [ Links ]
5. Fernandez–Ortiz A, Badimon JJ, Falk E et al: Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol 1994;23: 1562–1569. [ Links ]
6. Toschi V, Gallo R, Lettino M, Fallón JT, Fernandez–Ortiz A, Badimon L, et al. Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation 1997; 95: 594–599. [ Links ]
7. Levi M, Ten Cate H, Bauer KA, et al: Inhibition of endotoxin–induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal antitissue factor antibody in chimpanzees. J Clin Invest 1994; 93: 114–120. [ Links ]
8. Pixley RA, De LC, Page JD, et al: The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia: in vivo use of a monoclonal anti–factor XII antibody to block contact activation in baboons. J Clin Invest 1993, 91: 61–68 [ Links ]
9. Ruf W, Edgington TS: Structural biology of tissue factor, the initiator of thrombogenesis in vivo. FASEB J 1994; 8: 385–390. [ Links ]
10. Tedgui A, Mall at Z: Apoptosis as a determinant of atherothrombosis. Thromb Haemost 2001; 86: 420–426. [ Links ]
11. Cai W, Devaux B, Schaper W, et al: The role of Fas/APO 1 and apoptosis in the development of human atherosclerotic lesions. Atherosclerosis 1997; 131: 177–186. [ Links ]
12. Fadok VA, Bratton DL, Frasch SC, et al: The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ 1998; 5:551–562. [ Links ]
13. Mallat Z, Tedgui A: Current perspective on the role apoptosis in atherotrombotic disease. Circ Res 2001; 88: 998–1003. [ Links ]
14. Mallat Z, Hugel B, Ohan J, et al: Shed membrane microparticles with procoagulant potential inhuman atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 1999; 99: 348–353. [ Links ]
15. Levi M, Van der Poll T, Buller HR: Bidirectional relation between inflammation and coagulation. Circulation 2004; 109: 2698–2704. [ Links ]
16. Coughlin SR, Camerer E: Participation in inflammation. J Clin Invest 2003; 111: 25–27. [ Links ]
17. Coughlin SR. Thrombin signaling and proteases–activated. Nature 2000; 407: 258–264. [ Links ]
18. Sambrano GR, Weiss EJ, Zheng YW, et al: Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature 2001; 413: 74–78. [ Links ]
19. Marutsuka K, Hatakeyama K, Sato Y, et al: Protease–activated receptor 2 (PAR2) mediates vascular smooth muscle cell migration induced by tissue factor/factor VIIa complex. Thromb Res 2002; 107: 271–276. [ Links ]
20. Gerotziafas G, Zafiropoulos A, Van Dreden P, et al: Inhibition of factor VIIa generation and prothrombin activation by treatment with enoxaparin in patients with unstable. Br J Haematol 2003; 120: 611–617. [ Links ]
21. Keating FK, Harold HL, Whitaker DA, Sobel BE, Schneider DJ: The effects of bivalirudin compared with those of unfractionated heparin plus eptifíbatide on infíammation and thrombin generation and activity during coronary intervention. Coron Artery Dis 2005; 16: 401–405. [ Links ]

