










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.76 no.1 Ciudad de México ene./mar. 2006
Investigación clínica
Hipertensión arterial pulmonar en niños con síndrome de Down y cardiopatía congénita. ¿Es realmente más severa?
Pulmonary hypertension in children with Down's syndrome and congenital heart disease. Is it really more severe?
Clara A Vázquez–Antona,* Catalina Lomelí,** Alfonso Buendía,*** Jesús Vargas–Barrón*
* Departamento de Ecocardiografía. Instituto Nacional de Cardiología "Ignacio Chávez", México, D.F.
** Cardiología de Adultos. Instituto Nacional de Cardiología "Ignacio Chávez", México, D.F.
*** Departamento de Cardiología Pediátrica. Instituto Nacional de Cardiología "Ignacio Chávez", México, D.F.
Correspondencia:
Dra. Clara Andrea Vázquez Antona
Departamento de Ecocardiografía. Instituto Nacional de Cardiología "Ignacio Chávez"
(INCICH Juan Badiano Núm. 1. Col. Sección XVI, Tlalpan 14080 México, D.F).
Teléfono: (52) 5573–2911 ext. 1212 Fax: (52) 5573–0994.
Correo electrónico: cvazquezant@yahoo.com.mx
Recibido: 10 de enero 2006
Aceptado: 16 de enero 2006
Resumen
Objetivo: Comparar el comportamiento hemodinámico, la severidad y reversibilidad de la hipertensión arterial pulmonar en pacientes con síndrome de Down y cardiopatía congénita (CC) respecto a los sin cromosomopatía.
Material y métodos: Se estudiaron 30 pacientes con cardiopatía congénita acianógena y cortocircuito de izquierda a derecha, corroborados con ecocardiografía: 16 pacientes tenían síndrome de Down y CC y el grupo control estaba formado por 14 pacientes sin anormalidades cromosómicas y con CC. La edad fue de 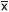 = 4.7 ± 5.8 años para el grupo con síndrome de Down y de = 5.3 ± 4.5 años para el grupo control. A todos se les realizó estudio hemodinámico completo, análisis estructural mediante angiografía pulmonar en cuña magnificada (APCM) y prueba con administración de oxígeno.
= 4.7 ± 5.8 años para el grupo con síndrome de Down y de = 5.3 ± 4.5 años para el grupo control. A todos se les realizó estudio hemodinámico completo, análisis estructural mediante angiografía pulmonar en cuña magnificada (APCM) y prueba con administración de oxígeno.
Resultados: El diagnóstico más frecuente fue comunicación interventricular para el grupo control y de canal AV común para el grupo Down. La presión pulmonar sistólica y media de la arteria pulmonar tuvieron valores muy similares en ambos grupos, con un promedio de 84.87 ± 13.16 mm Hg para la presión sistólica en el grupo Down, y de 84.21 ± 22.05 mm Hg para el grupo control. Posteriormente a la aplicación de oxígeno existió una tendencia de incremento en la relación Qp/Qs con la caída de las resistencias vasculares pulmonares en ambos grupos, siendo más importante en el grupo control. Durante la valoración mediante APCM no se observaron diferencias en la valoración cuantitativa en ambos grupos, ni posterior a la angiografía con oxígeno.
Conclusiones: A pesar que los pacientes con síndrome de Down presentan cardiopatías con mayor predisposición a desarrollar hipertensión arterial pulmonar irreversible como el canal atrioventricular común, el comportamiento hemodinámico de la HAP y durante el reto con oxígeno fue similar en ambos grupos.
Palabras clave: Síndrome de Down. Hipertensión pulmonar. Angiografía pulmonar en cuña. Cardiopatías congénitas.
Summary
Objective: To compare the hemodynamic state, the severity and reversibility of pulmonary arterial hypertension (PAH) in patients with Down's syndrome and congenital heart disease (CHD) with respect to those without chromosomal pathologies.
Material and methods: 30 patients with congenital heart disease and left to right shunt were studied, corroborated by echocardiography; 16 patients had Down's syndrome and CHD and the control group was constituted by 14 patients without chromosomal abnormalities and with CHD. The age was = 4.7 ± 5.8 years for the Down's syndrome group and x = 5.3 ± 4.5 years for the control group. All patients were subjected to a complete hemodynamic study, as well as to structural analysis by pulmonary wedge angiography (PWA), tested with oxygen administration.
Results: The most frequent diagnosis was ventricular septal defect for the control group and common atrioventricular canal for the Down's group. The systolic and mean pulmonary pressure depicted very similar values in both groups, with an average of 84.87 ± 13.16 mm Hg for the Down's group and 84.21 ± 22.05 for the control group. After oxygen administration, a tendency of increased Qp/Qs was found with a drop in pulmonary resistance in both groups, but being more important in the control group. During PWA assessment no quantitative differences were observed in PAH between both groups nor after the angiography with oxygen administration.
Conclusions: Although patients with Down's syndrome present CHD with greater predisposition to develop irreversible pulmonary arterial hypertension like common atrioventricular canal, the hemodynamic behavior of pulmonary hypertension and during the challenge with oxygen was similar in both groups.
Key words: Down's syndrome. Congenital heart disease. Pulmonary wedge angiography. Hypertensive pulmonary vascular disease.
Introducción
El síndrome de Down es la anormalidad cromosómica más frecuente, se presenta en uno de cada 800 nacidos vivos.1 Aproximadamente el 40% de niños con este síndrome tienen alguna cardiopatía o malformación cardíaca congénita,2 esta asociación se incrementa hasta el 62% cuando el análisis se realiza en pacientes hospitalizados.3 La malformación cardíaca que más se asocia es el defecto de la tabicación atrioventricular (canal AV).4 En nuestro medio la incidencia entre síndrome de Down y cardiopatía congénita varía del 36%5 al 58%,6 siendo la lesión cardíaca más frecuente sola o asociada a otra cardiopatía, el conducto arterioso permeable (PCA), seguida por la comunicación interventricular (CIV) e interauricular (CIA). A diferencia de lo publicado en otros países, el canal AV sólo se reporta en cerca del 10% de los casos. Otras malformaciones que se presentan con menor frecuencia son la Tetralogía de Fallot, coartación aórtica y atresia tricuspídea.
Varios autores sugieren que los niños con síndrome de Down pueden desarrollar cambios en la vasculatura pulmonar en forma más temprana y que son rápidamente progresivos a diferencia de niños con cromosomas normales y que tienen defectos cardíacos congénitos similares,7,8sin embargo otros autores como Plett y Wilson9,10 indican que si bien los niños con síndrome de Down tienen una alta incidencia de hipertensión arterial pulmonar (HAP), esta hipertensión no es más severa que la observada en pacientes sin este síndrome.
Se ha considerado que la mejor forma de valorar la severidad y reversibilidad de la HAP es mediante el cateterismo cardíaco a través de las mediciones hemodinámicas y el análisis estructural con la angiografía pulmonar en cuña magnificada (APCM), la cual valora los cambios estructurales de la vasculatura pulmonar basándose tanto en mediciones como en la morfología de las fases arterial, capilar y venosa.11
El objetivo de este estudio fue comparar el comportamiento hemodinámico, la severidad y reversibilidad de la HAP en pacientes con síndrome de Down y cardiopatía congénita de aquéllos sin cromosomopatía, y su correlación con la valoración de la anatomía vascular pulmonar analizada mediante angiografía pulmonar en cuña magnificada.
Material y métodos
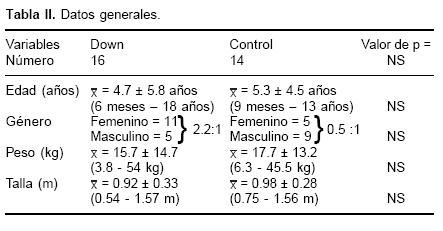
Se estudiaron 30 pacientes hospitalizados en el servicio de Cardiología Pediátrica del Instituto Nacional de Cardiología "Ignacio Chávez", con cardiopatía congénita acianógena y cortocircuito de izquierda a derecha, corroborado por clínica y ecocardiografía. Los pacientes fueron divididos en dos grupos: el primero, formado por 16 pacientes portadores del síndrome de Down y cardiopatía congénita acianógena y el grupo control formado por 14 pacientes sin anormalidades cromosómicas y con cardiopatía congénita. El rango de edad fue de 6 meses a 18 años para el grupo con síndrome de Down y de 9 meses a 13 años para el grupo control. Fueron excluidos los enfermos cuya evolución fue desconocida.
A todos los pacientes se les elaboró historia clínica completa, examen físico, radiografía de tórax, electrocardiograma y ecocardiograma como parte del abordaje diagnóstico, posteriormente se realizó estudio hemodinámico. El protocolo al que fueron sometidos se dividió en 3 fases:
1. Estudio hemodinámico completo de acuerdo con el tipo de cardiopatía con mediciones al aire ambiente.
2. Análisis estructural mediante APCM.
3. Prueba con vasodilatadores pulmonares (administración de oxígeno) con valoración hemodinámica y de APCM.
1. Análisis hemodinámico.
En la primera fase del estudio hemodinámico se realizaron las mediciones al aire ambiente, se colocaron catéteres venoso y arterial (se administró heparina a la dosis de 50 a 100 unidades por kilogramo de peso). Posteriormente se tomaron muestras para oximetrías de acuerdo al tipo de cardiopatía para determinar los saltos volumétricos y la presencia de cortocircuitos. Se realizó registro de presiones para obtener posteriormente los siguientes cálculos hemodinámicos para valorar HAP:
• Gasto pulmonar total (GPT).
• Gasto pulmonar efectivo (GPe).
• Resistencias vasculares pulmonares y resistencias pulmonares totales (Rvp y Rpt) en unidades Wood/m2.
• Resistencias vasculares sistémicas (Rvs).
• Relación Rvp/Rvs.
• Relación PAP/PAo.
Para el cálculo de gastos se utilizó el método de Fick con el consumo de oxígeno (VO2) calculado,12 conociendo que en el cálculo pueden influir varios factores como la sedación, hematócrito, colaterales, etc. Las resistencias vasculares fueron medidas en unidades Wood/m2.
2. Análisis estructural mediante APCM.
Se utilizó la técnica de enclavamiento directo en la arteria pulmonar con catéter de Goodlabe Lubin. El sitio de colocación es el lóbulo inferior derecho o izquierdo. Se inyectó 0.5 a 3 mL de medio de contraste para valorar el segmento de la vasculatura pulmonar que fue aislado. El método de corrección de magnificación fue posible porque el nivel de la tabla de rayos X fue modificada, el tubo fue separado 1.50 m de la mesa utilizando de 60 a 70 Kv. Para interpretar la cineangiografía el tagarno fue separado de 1.8 a 2 metros de la pantalla, con lo que se obtiene una magnificación de 7 a 8 veces.13
La interpretación de la APCM se basó en datos objetivos como es el caso de las mediciones de arterias proximales, venas paralobulares, arterias monopediales y tiempo de llenado capilar y datos subjetivos que valoraron la morfología de la vasculatura pulmonar de fase arterial, de llenado capilar y venosa (Fig. 1). La fase arterial se valoró con el llenado de las arterias paralobulares seguida por las arterias monopediales o vasos supernumerarios que son aquellos vasos que nacen de las arterias musculares o paralobulares sobre el ángulo derecho (la cuenta fue realizada a una distancia de 10 mm de las arterias musculares). La mancha capilar normal debe ser homogénea y completa además de rodear a todas las arterias distales de los lóbulos. Finalmente fue valorada la fase venosa y el tiempo de llenado capilar que es desde que inicia la mancha capilar hasta que aparece la primera vena drenando al atrio izquierdo hacia el ángulo izquierdo. En la morfología normal la fase arterial presenta un llenado temprano de la vena paralobular. La valoración de las arterias están en relación a su tamaño, disminución gradual del calibre y arborización. De acuerdo a estos datos la valoración morfológica de la angiografía pulmonar en cuña se clasifica en normal, regular y mala anatomía (Tabla I).

Las mediciones que se realizaron y que ya han sido establecidas previamente fueron las siguientes (Fig. 2):14
• Relación vena/arteria (normal: 1/1)
• Diámetro de las arterias monopediales (> 300 mieras).
• La amputación o corte temprano de las arterias (la longitud de un vaso es de 1.5 a 2.5 mm).
• Tiempo de circulación pulmonar (normal: < 0.6 segundos).
3. Prueba con vasodilatadores pulmonares.
La última fase del estudio corresponde a la prueba de reactividad pulmonar con la aplicación de oxígeno como vasodilatador pulmonar. Se colocó al paciente una mascarilla con O2 al 100% durante 10 minutos y se tomaron nuevamente muestras para oximetrías y registro de presiones con una nueva medición de los cálculos hemodinámicos. Finalmente se realizó una nueva APCM con nuevas mediciones y análisis morfológico.
Análisis estadístico
Para el análisis estadístico se utilizó la prueba de rangos señalados y pares igualados de Wilcoxon, posteriormente con la administración de O2 debido a la reducción del número de pacientes con cada grupo se utilizó la prueba de U de Mann–Whitney que es una prueba no paramétrica para muestras pequeñas.
Resultados
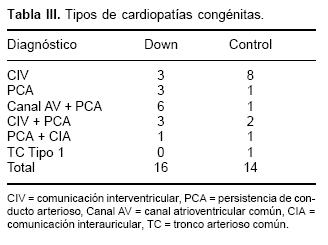
Ambos grupos fueron similares en relación con la edad, peso y talla, sin encontrar una diferencia estadísticamente significativa (p < de 0.05) (Tabla II). Sólo en el grupo de Down se encontró como hallazgo predominio del sexo femenino. En relación con el diagnóstico la mayor frecuencia fue de CIV para el grupo control y de canal AV (de estos cuatro pacientes además con PCA) para el grupo Down (Tabla III). En ambos grupos la HAP fue severa.
Valoración hemodinámica
Sin oxígeno. No se encontró diferencia estadísticamente significativa durante la valoración hemodinámica de la HAP, de tal manera que el comportamiento fue de la misma severidad en ambos grupos. Únicamente en relación al flujo sistémico y pulmonar, se observó mayor flujo pulmonar en el grupo control, traducido por una Qp/Qs mayor de 1.5 (p = 0.02). La presión pulmonar sistólica y media de la arteria pulmonar tuvieron valores muy similares en ambos grupos, mostrando HAP severa (Tabla IV). A pesar que el valor de las Rvp fue mayor para el grupo Down que para el grupo control (13.11 + 11.5 U/m2 vs 9.25 + 5.4 U/m2) no fue estadísticamente significativo. La relación entre las resistencias vasculares pulmonares y sistémicas fue ligeramente mayor en el grupo con síndrome de Down, sin que mostrara significancia estadística.
Con aplicación de oxígeno. La valoración hemodinámica con la administración de oxígeno se realizó sólo en 11 pacientes del grupo Down y en 13 pacientes del grupo control (Tabla IV), este factor fue tomado en cuenta al realizar el análisis estadístico. El comportamiento hemodinámico fue similar en ambos grupos. Se observó una caída de las resistencias vasculares pulmonares con incremento del Qp/Qs en el grupo control, sin embargo al valorar la relación entre tres resistencias pulmonares y sistémicas y entre presión pulmonar y aórtica no se observaron cambios en relación a los pacientes con síndrome de Down.
Angiografía pulmonar en cuña magnificada.
Valoración cuantitativa
Sin oxígeno. En la valoración angiográfica sin la aplicación de oxígeno se observó que las arterias proximales fueron del mismo diámetro en ambos grupos; la relación V/A fue de 1.1:1 para el grupo Down y para el grupo control. Las arterias monopediales mostraron en ambos grupos un valor superior a 600 mieras con un tiempo de llenado capilar de 0.9 segundos para el grupo Down y de 1.1 segundos para el grupo control (Tabla V). Estos datos concuerdan con los datos hemodinámicos que indican HAP severa en ambos grupos.
Con aplicación de oxígeno. Durante la valoración con oxígeno se observó en el grupo Down una discreta disminución del calibre de las arterias monopediales, no así para el grupo control, sin embargo no se encontró significancia estadística, lo mismo para el tiempo de llenado capilar que disminuyó de 0.9 a 0.7 en el grupo Down y de 1.1 a 0 en el grupo control (Tabla V). Al analizar el comportamiento después de la aplicación con O2 no existió diferencia entre ambos grupos.
Valoración morfológica
Grupo control. Se analizó la morfología de la vasculatura pulmonar en base a los parámetros previamente citados sólo en 10 pacientes del grupo control (Fig. 3); sin administración de O2 se observó anatomía normal sólo en uno, regular en 2 y mala anatomía en 7; con administración de O2 los pacientes con anatomía regular mejoraron mostrando anatomía normal, no así los pacientes con mala anatomía ya que sólo 3 mejoraron su anatomía considerándose regular después de la administración de O2.

Al correlacionar los hallazgos morfológicos con los parámetros hemodinámicos se observó que el paciente que presentó morfología regular que mejora con oxígeno, la HAP era moderada con caída importante de las resistencias vasculares pulmonares.
De los pacientes con mala anatomía que mejoran a anatomía regular posterior a la aplicación de oxígeno, en dos de ellos se observó caída de las Rvp y el otro persistió con Rvp altas.
Los seis pacientes que mostraron mejoría de la APCM posterior a la aplicación de oxígeno fueron llevados a tratamiento quirúrgico con adecuada evolución y regresión de la presión pulmonar a valores normales, a excepción del paciente con anatomía regular y RVP que persistieron altas, quien se mantiene con HAP moderada a 10 años de la cirugía.
Los diagnósticos de los cuatro pacientes con mala anatomía y que no presentaron cambios fueron: tronco arterioso tipo I, canal atrioventricular común y dos pacientes con CIV amplia. Todos tenían HAP severa con PSAP entre 79 y 140 mm Hg.
Los datos generales de la valoración hemodinámica y angiográfica así como del tratamiento y evolución se muestran en la Tabla VI.
Grupo Down. La morfología de la vasculatura pulmonar se analizó en 13 pacientes de este grupo (Fig. 4). Tres de ellos presentaron buena anatomía antes y después de la administración de O2 y fueron llevados a cirugía; estos pacientes presentaron PSAP entre 60 y 75 mm Hg y Rvp entre 4 y 11 UW/m2 que disminuyeron posterior a la aplicación de oxígeno y fueron llevados a tratamiento quirúrgico. Cinco pacientes presentaron regular anatomía y posterior a la administración de O2, sólo en 4 mejoró la anatomía. La PSAP de estos pacientes varió de 40 a 100 mm Hg, todos tenían Rvp mayores de 10 U/m2 con caída posterior a la aplicación de O2. Fueron llevados a tratamiento quirúrgico con manejo para HAP en el postoperatorio temprano y regresión de la HAP; un paciente falleció por sepsis. El paciente con anatomía regular que no presentó respuesta a la administración de O2 se consideró fuera de tratamiento quirúrgico.

En cinco pacientes se observó angiografía pulmonar en cuña con mala anatomía, de los cuales sólo dos mostraron respuesta satisfactoria a la administración de O2, uno de ellos pasó a regular y otro a buena anatomía (Fig. 5A) y fueron sometidos a tratamiento quirúrgico, ambos pacientes presentaron en el postoperatorio crisis hipertensivas manejados con vasodilatadores, con evolución postoperatoria adecuada, manteniendo durante el seguimiento PSAP alrededor de 40 mm Hg, los diagnósticos eran de CIV y CIA más PCA (Tabla VII).
Tres pacientes con mala anatomía permanecieron sin cambios después de la administración de O2 y fueron considerados fuera de tratamiento quirúrgico (Fig. 5B). Los diagnósticos que presentaban estos pacientes fueron: Canal AV compun, PCA y el tercero CIV más PCA. Las Rvp de 32, 46 y 20 UW/m2 respectivamente, con PSAP entre 90 y 100 mm Hg.
En este grupo en general se observó que el 69% presentó mejoría de la morfología posterior a la aplicación de O2, considerándose sólo el 31% fuera de tratamiento quirúrgico mientras que en el grupo control la respuesta a la administración de O2 fue del 60% y se consideró fuera de tratamiento quirúrgico el 40%.
Discusión
Las implicaciones quirúrgicas de hipertensión arterial pulmonar (HAP) y enfermedad vascular pulmonar obstructiva (EVPO) secundaria a cardiopatías congénitas fueron reconocidas a principios de los años 50.15,16 En 1958 Heath and Edwards17 realizaron la descripción patológica de los cambios vasculares pulmonares que ocurren en pacientes con defectos septales congénitos y presión arterial pulmonar elevada. A principios de la década de los 60 De Micheli y cols18 establecen que las alteraciones histológicas de la capa media, en presencia de hipertensión arterial moderada y de la capa media e íntima de las arterias pulmonares en presencia de hipertensión severa, son producidas por las condiciones circulatorias particulares del lecho vascular pulmonar. Durante los pasados 30 años, cardiólogos y cirujanos realizaron pruebas para determinar la edad óptima de intervención quirúrgica y evitar el desarrollo de hipertensión arterial pulmonar. Inicialmente los datos obtenidos sugerían que los cambios vasculares pulmonares eran generalmente reversibles dentro de los primeros 24 meses de vida, pero estudios más recientes han confirmado que estos cambios pueden ocurrir incluso dentro de los primeros 12 meses de edad, particularmente con canal aurículo–ventricular común.19,20
La valoración hemodinámica preoperatoria de la hipertensión arterial pulmonar es importante en muchos tipos de cardiopatías congénitas; los signos y síntomas que se presentan en ocasiones no se correlacionan con el grado de enfermedad vascular pulmonar y es clínicamente difícil definir en qué pacientes se presentará mejoría de la HAP posterior al tratamiento quirúrgico.
El cateterismo cardíaco es considerado como el mejor método para valorar el comportamiento hemodinámico de la HAP y las resistencias vasculares pulmonares a pesar de que tiene limitaciones. Una de ellas es en relación al cálculo teórico del consumo de oxígeno utilizado en el cálculo de gastos pulmonares y sistémicos, además del uso de la fórmula de Poiseuille's cuando se calculan las resistencias vasculares pulmonares.12,21 La biopsia pulmonar también tiene un papel importante, sin embargo algunos grupos han fundamentado una pobre correlación entre los estudios hemodinámicos y los cambios de la estructura pulmonar, además de ser un procedimiento quirúrgico que presenta complicaciones y puede no ser predictor de operabilidad.19,22
Es conocido que en respuesta a flujo y presiones elevadas secundarias a cortocircuito arteriovenoso por cardiopatía congénita ocurren anormalidades estructurales progresivas en el lecho vascular pulmonar que modifican la morfología normal, estas alteraciones pueden ser observadas a través del análisis morfológico y cuantitativo de la APCM. Como ha sido puntualizado por Hoffman22 y Rabinovitch,14es de considerable valor si el estado estructural de la vasculatura pulmonar es valorada al mismo tiempo del cateterismo cardíaco con la finalidad de identificar aquellos casos con enfermedad vascular pulmonar obstructiva (EVPO) que pueden permanecer sin cambios o incluso empeorar y que no son candidatos a tratamiento quirúrgico, de aquellos que pueden obtener el beneficio de la cirugía con regresión de la HAP y EVPO.13,14,23–25 Wilson y cols.10 demostraron que cuando los parámetros hemodinámicos y los obtenidos por APCM son concordantes, los cambios estructurales de la enfermedad vascular pulmonar son precisos y pueden considerarse como un importante predictor de la enfermedad sin necesidad de que se realice biopsia pulmonar. Los datos morfológicos de mayor importancia en la valoración son la disminución gradual de las arterias, una adecuada arborización y la mancha capilar. Cuando las arterias muestran amputación temprana, poca arborización y una mancha capilar incompleta (mala anatomía) estos hallazgos tienen una adecuada correlación con resistencias pulmonares elevadas. Ravitnovich26 en uno de sus estudios iniciales clasifica los hallazgos de la APCM en tres grupos, llamado el "ABC" de la hipertensión pulmonar, en donde correlaciona los hallazgos patológicos de la APCM con el flujo, presión y resistencias pulmonares. En el presente trabajo no observamos, como previamente se ha reportado, una correlación adecuada entre presión y resistencias pulmonares con los hallazgos morfológicos de la APCM, ya que por los valores hemodinámicos encontrados todos los pacientes deberían tener mala anatomía; observamos que en la anatomía vascular pulmonar normal las resistencias vasculares pulmonares fueron menores a 6U/m2, mientras que en la anatomía regular las Rvp variaron entre 6 y 10 U/m2, sin embargo en la "mala" anatomía no existió en la mayoría de los casos concordancia con la valoración hemodinámica.
El reto con vasodilatadores pulmonares, en este caso el oxígeno, ayuda a definir cuándo estos cambios pueden ser reactivos y por lo tanto reversibles como se muestra en el presente trabajo, en donde se observa mejoría de la morfología de la vasculatura pulmonar en aproximadamente la mitad de los casos de ambos grupos, por lo que pueden ser llevados a tratamiento quirúrgico pacientes que inicialmente eran considerados fuera de cirugía. A excepción de un paciente que persistió con HAP durante el seguimiento, en el resto de los pacientes la evolución fue adecuada, con regresión de la HAP. La experiencia con nuestro grupo de trabajo apoya estas observaciones,11 considerando que la APCM ayuda a predecir qué pacientes tendrán reversibilidad de los cambios en la vasculatura producidos por la HAP y la evolución postoperatoria, definiendo además qué pacientes pueden ser susceptibles de presentar crisis hipertensivas pulmonares en el postoperatorio inmediato.
Es conocido que entre más compleja sea la cardiopatía o más importante el cortocircuito arteriovenoso inicial, los pacientes tienen mayor posibilidad de presentar EVPO que los convierte en inoperables a menor edad, como en el defecto del tabique atrioventricular que durante la infancia tienen resistencias altas, flujo hemodinámico bajo y avanzada enfermedad a nivel de la estructura vascular pulmonar.20 Un hecho importante en este trabajo es que la HAP encontrada en ambos grupos fue severa, independientemente del tipo de defecto, consideramos que estos resultados representan la evolución natural de la HAP en pacientes mayores.
Por otro lado, los pacientes con síndrome de Down desarrollan en forma precoz HAP, aun en ausencia de cardiopatía congénita y se han emitido algunas hipótesis para explicarla. Cooney y Thurlbeck27 demostraron la presencia de anormalidades en el desarrollo pulmonar con disminución en el número de alvéolos en relación a los ácinos y un área de superficie alveolar pequeña, lo que acompañada de una pérdida de capilares es responsable del agravamiento de la HAP en estos pacientes. También presentan disfunción de la ventilación y obstrucción de las vías aéreas superiores secundaria a adenoides, laringomalacia, apnea obstructiva del sueño, hipertrofia de la lengua y macizo facial pequeño que causan hipoxemia crónica, lo que aunado a la hipotonía con la que cursan estos pacientes provoca la posible ausencia de involución del patrón vascular fetal y evolución directa a la EVPO. Otros autores como Yamaki y cols28 demostraron que los pacientes con síndrome de Down tienen además adelgazamiento de la capa media de las arterias, lo que favorece la proliferación fibrosa de la capa íntima. Además se ha observado que cuando estos pacientes tienen cardiopatía presentan asociación a PCA con mayor frecuencia, lo que también predispone a desarrollo precoz de la HAP y a mayor severidad.10 Esta asociación de cardiopatía y PCA también se demuestra en el presente trabajo.
Nosotros observamos que a pesar de que en el grupo con síndrome de Down se presentan cardiopatías con mayor predisposición a desarrollar hipertensión arterial pulmonar irreversible como el canal atrioventricular común, el comportamiento hemodinámico de la HAP fue similar a los niños con cromosomas normales, a diferencia de lo reportado por Clapp y cols,29 quienes reportan que los niños con síndrome de Down y canal AV tienen una elevación mayor de las Rvp y rápida progresión de la EVPO que los niños con cromosomas normales. Los hallazgos en relación a una mejor respuesta a la aplicación de oxígeno del diámetro de las arterias monopediales en el grupo Down, hace suponer que estos pacientes pudieran presentar un componente más reactivo. Estos hallazgos pueden ser importantes en la clínica y en el manejo quirúrgico, tomando en cuenta que cuando son sometidos a cirugía en el momento oportuno su evolución es satisfactoria, sin embargo es importante tener presente que las complicaciones extracardíacas pueden presentarse con mayor frecuencia en los pacientes con esta cromosomopatía y ocasionar mala evolución,30 como sucedió con dos pacientes que fallecieron en el perioperatorio por complicaciones infecciosas.
Es importante mencionar que en ambos grupos la valoración cuantitativa y morfológica de la APCM fue un parámetro determinante como predictor de enfermedad vascular pulmonar.
Referencias
1. Baird PA, Sadovnick AD: Life tables for Down Syndrome. Hum Genet 1989; 82: 291–292. [ Links ]
2. Korenberg J, Kurnit D: Molecular and stochastic basis of congenital heart defects in Down Syndrome. En: Marino B, Pueschel SM. "Heart disease in persons with Down syndrome". Baltimore: Brookes, 1996: 21–38. [ Links ]
3. Greenwood RD, Nadas AS: The Clinical Course of Cardiac Disease. En: Down's Syndrome. Pediatrics 1976; 58: 893–897. [ Links ]
4. Marino B: Patterns of congenital heart disease and associated cardiac anomalies in children with Down syndrome. En: Marino B, Pueschel SM. "Heart disease in persons with Down's syndrome". Baltimore: Brookes, 1996: 133–40. [ Links ]
5. Rodríguez–Hernández L, Reyes–Nuñez J: Cardiopatías congénitas en el síndrome de Down. Bol Méd Hosp Infant Mex 1984; 41: 622–625. [ Links ]
6. De Rubens FJ, Del Pozzo MB, Pablos JL, Calderón C, Castrejón R: Malformaciones cardíacas en los niños con síndrome de Down. Rev Esp Cardiol 2003; 56: 894–899. [ Links ]
7. Laursen HB: Congenital heart disease in Down's syndrome. Br Heart J 1976; 39: 721–726. [ Links ]
8. Chi TL, Krovetz JL: The pulmonary vascular bed in children with Down's syndrome. J Pediatr 1975; 86: 533–538. [ Links ]
9. Plett JA, Tandon R, Moller JH, Edwards JE: Hypertensive Pulmonary Vascular Disease. Arch Pathol 1974; 97: 187–188. [ Links ]
10. Wilson SK, Grover MH, Nelly CA: Hypertensive pulmonary vascular disease in Down's syndrome. J Pediat 1979; 95: 722–726. [ Links ]
11. Vázquez Antona C, Calderón J, Gil M, García y Otero A, Buendía A, Martínez M, Attie F: Estudio en pacientes con hipertensión arterial secundaria a cardiopatía congénita mediante angiografía pulmonar en cuña magnificada. Arch Inst Cardiol Mex 1991; 61(4) supl: 4. [ Links ]
12. La Farge CG, Miettinem OS: The estimulation of oxygen consumption. Cardiovasc Res 1970; 4: 23–27. [ Links ]
13. Nihill MR, McNamara DG: Magnification pulmonary wedge angiography in the evaluation of children with congenital heart disease and pulmonary hypertension. Circulation 1978; 58–6: 1094–1106. [ Links ]
14. Rabinovitch M, Keane JF, Fellows EK, Castañeda AR, Reid L: Quantitative analysis of the pulmonary wedge angiogram in congenital heart defects. Circulation 1981; 63: 152–164. [ Links ]
15. Civin WB, Edwards JE: Pathology of the pulmonary vascular tree. A comparison of intrapulmonary arteries in Eisenmenger's complex and in stenosis of ostium infudibuli associated with biventricular origin of the aorta. Circulation 1950; 2: 545–553. [ Links ]
16. Selzer A, Laqueur GL: The Eisenmenger complex and its relation to uncomplicated defect of the ventricular septum. Arch Intern Med 1951; 87: 218–226. [ Links ]
17. Heath D, Edwards JE: The pathology of hypertensive pulmonary vascular disease: a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 1958; 18: 533–547. [ Links ]
18. De Michelli A, Piccolo E, Espino Vela J, Monroy G, Rubio Alvarez V: Observaciones sobre los mecanismos reguladores de las presiones pulmonares en las cardiopatías congénitas con cortocircuito arteriovenoso. Arch Inst Cardiol Mex 1960; 30: 527–555. [ Links ]
19. Dammann JF, Ferencz C: The significance of the pulmonary vascular bed in congenital heart disease. III Defects between the ventricles or great vessels in which both increased pressure and blood flow may act upon the lungs and in which there is a common ejective force. Am Heart J 1956; 52: 210–231. [ Links ]
20. Newfeld EA, Sher M, Paul MH, Nikaidoh H: Pulmonary vascular disease in complete atrioventricular canal defect. Am J Cardiol 1977; 39: 721–726. [ Links ]
21. Frescura C, Thiene G, Franceschini E, Talenti E, Mazzuco A: Pulmonary vascular disease in infants with complete atrioventricular septal defect. Int J Cardiol 1987; 15: 91–100. [ Links ]
22. Hoffman JIE: Diagnosis and treatment of pulmonary vascular disease. Birth Defects 1972; 8:9–18. [ Links ]
23. Fried R, Falkovski G, Newburger J, Gorchacova AI, Rabitnovitch M, Gordonova M, et al: Pulmonary arterial changes in patients with ventricular septal defects and severe pulmonary hypertension. Pediatr Cardiol 1986; 7: 147–154. [ Links ]
24. Castellanos A, Hernández FA, Mercado HG: Wedge pulmonary arteriography in congenital heart disease. Radiology 1965; 85: 838–849. [ Links ]
25. Fujiyama J, Yoshida Y, Ogata H, Kato T, Kuteken O, Oyama K: Pulmonary wedge angiography for the evaluation of the pulmonary vascular bed in congenital heart disease. Cath Cardiovasc Diag 1984; 10: 237–246. [ Links ]
26. Ravitnovitch M, Haworth S, Castañeda A, Nadas A, Reid L. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 1978; 58:1107–1122. [ Links ]
27. Cooney T, Thurlbeck W: Pulmonary hypoplasia in Down's Syndrome. N Engl J Med 1982; 307: 1170–1173. [ Links ]
28. Yamaki S, Horiuchi T, Sekino Y: Quantitative analysis of pulmonary vascular disease in simple cardiac anomalies with the Down's Syndrome. Am J Cardiol 1983; 51: 1502–1506. [ Links ]
29. Clapp S, Perry BL, Farooki ZQ, Jackson WL, Karpawich PP, Hakimi M, et al: Down's syndrome, complete atrioventricular canal, and pulmonary vascular obstructive disease. J Thorac Cardiovasc Surg 1990; 100: 115–21. [ Links ]
30. Calderón–Colmenero J, Flores A, Ramírez S, Patiño–Bahena E, Zabal C, García–Montes JA, y cols: Resultados en la corrección quirúrgica de la cardiopatía congénita en el síndrome de Down. Arch Card Mex 2004; 74: 39–44. [ Links ]

