










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.75 supl.3 Ciudad de México jul./sep. 2005
Comunicaciones breves
Rabdomioma cardiaco tratado quirúrgicamente con éxito y revisión de la literatura
Cardiac rhabdomyoma surgically treated with success. Review of literature
José Ángel Cigarroa López,*Yoloxóchitl García Jiménez,* Lucelly Yáñez Gutiérrez,* Santiago Jiménez Arteaga,* Arturo Martínez Sánchez,* José Ortegón Cardeña,* Felipe David Gómez,* Agustín Sánchez Soberanes,* Diana López Gallegos,* Carlos Riera–Kinkel,* Carlos Alva Espinosa*
* Servicio de Cardiopatías Congénitas, Hospital de Cardiología; Centro Médico Nacional Siglo XXI.
Correspondencia:
José Ángel Cigarroa López.
Hospital de Cardiología, CMN Siglo XXI.
Cuauhtemoc Num. 330, Col. Doctores, Delegación Cuauhtemoc; México, Distrito Federal.
Tel: 56276900 Ext: 22203.
Correo electrónico: ciga09@prodigy.net.mx
Resumen
Los tumores cardiacos primarios son raros, con incidencia variable en todas las edades del 0.005 al 0.05%. En pacientes pediátricos, la incidencia es del 0.27%. Los tumores más frecuentes durante la infancia son los rabdomiomas cardiacos, considerados como benignos. Aunque la expresión clínica es amplia, en la mayoría de los casos son asintomáticos y se detectan por la presencia de soplos. En la etapa prenatal se manifiestan con arritmias o hydrops fetalis. En algunos neonatos y lactantes se encuentran arritmias, datos de bajo gasto cardíaco o muerte súbita. La asociación con esclerosis tuberosa se ha observado hasta en un 81%. Se presenta el caso de paciente masculino neonato, con diagnóstico de rabdomioma cardiaco que inicialmente estaba asintomático, sin embargo en el seguimiento requirió de tratamiento quirúrgico a los 5 meses de edad, por datos de insuficiencia cardiaca secundaria a obstrucción del tracto de salida del ventrículo derecho. A 5 meses de la cirugía, el paciente está asintomático.
Palabras clave: Tumor cardiaco. Rabdomioma. Esclerosis tuberosa.
Summary
The primary cardiac tumors are inusual, the incidence varies in all the ages between 0.005 to 0.05%. In pediatrics patients the incidence is 0.27%. The more frequent tumors during the childhood are the cardiac rhabdomyomas. These tumors are considered benigns. The clinical expression is wide, in the most the cases, the patients are asymptomatic and are detected by murmurs. In the prenatal age are manifested by arrhythmias or hydrops fetalis. The neonates and children may be show cardiac arrhythmias, low cardiac index and sudden cardiac death. The association with tuberous sclerosis had been reported in 81%.We presenta neonate with cardiac rhabdomyoma diagnosed in the newborn period when he was asymptomatic, however in the follow–up he developed cardiac failure by obstruction in the out flow tract of the right ventricle. He underwent open cardiac surgery to resect the obstruction. Five months after surgery, the patient remain asymptomatic.
Key words: Heart tumor. Rhabdomyoma. Tuberous sclerosis.
Introducción
Los tumores del corazón, son entidades raras en la práctica médica. Lymburner encontró una incidencia del 0.05% en 8,500 autopsias. En varias series de autopsias, la incidencia registrada es del 0.027 al 0.08% en pacientes pediátricos y la prevalencia de 1:10,000.1,2
El 90% de los tumores cardíacos primarios son benignos en la edad pediátrica. Sin embargo, pueden tener un comportamiento desfavorable, dependiendo de su localización, tamaño y número de tumores. La forma histológica más común es el rabdomioma en la etapa fetal y pediátrica. El rabdomioma fue descrito por primera vez por von Recklinghausenen 1862 asociado a neurofibromatosis.3 Se caracteriza por ser circunscrito, lobulado, blanquecino o grisáceo, habitualmente múltiple hasta en el 90% de los casos y generalmente se encuentra en el espesor del músculo cardiaco, pudiendo afectar cualquier punto, predominando en ventrículos y de éstos, el ventrículo izquierdo.4
El rabdomioma puede hallarse comúnmente asociado a esclerosis tuberosa hasta en el 81.1% de los casos,5 sin embargo, Tworetzky et al reportan 95% en una serie de 64 fetos y neonatos.2 Puede encontrarse solo o asociado a otras malformaciones congénitas. El ecocardiograma es la primera modalidad diagnóstica, reportándose el primer caso por ecocardiografia en 1982 por De Vore en etapa prenatal. Desde entonces se ha reportado intrauterinamente, una incidencia de 0.009% en la población abierta y 0.2% para los centros de referencia.5,6
El diagnóstico prenatal de rabdomioma se realiza principalmente durante el ultrasonido obstétrico de rutina, pudiendo ser referido por hallazgo de tumoración fetal cardiaca, arritmias o hydrops. Cuando el diagnóstico se realiza en etapa postnatal, es por escrutinio en pacientes con signos y síntomas de esclerosis tuberosa, cuando hay historia familiar o detección de soplo cardiaco.6,7 Menos frecuente, cuando se presentan síntomas cardiacos que requieran de tratamiento médico o quirúrgico.8 En pacientes asintomáticos sin datos de esclerosis tuberosa, pueden pasar inadvertidos, por lo cual la incidencia real de rabdomioma en niños y la frecuencia de la asociación con esclerosis tuberosa es incierta.9–12
Se ha descrito regresión parcial o total espontánea, hasta en el 80% de los casos, sin embargo, Fesslova y cols, reportan regresión parcial o total del 100% en una serie de 9 pacientes.5
La esclerosis tuberosa o enfermedad de Bourneville es un síndrome neurocutáneo, que se transmite en forma autosómica dominante, caracterizada por desarrollo de lesiones hamartomatosas en múltiples órganos, predominando en el cerebro, con tuberosidades corticales y nodulos subependimarios. Puede haber angiomiolipomas renales, hamartomas retiñíanos, linfangiomatosis pulmonar, lesiones cutáneas. En el 90% se presenta por mutaciones de novo, detectándose estas mutaciones en el cromosoma 9q (TSC–1) y en el 16p (TSC–2).2,9,12,13 El diagnóstico puede sospecharse mediante la tríada de Vogt que consiste en retraso mental, convulsiones y lesiones cutáneas. Sin embargo, existen criterios establecidos en 1992 y modificados en 1998 para el diagnóstico de esta entidad.3,6,12,14
El objetivo de este trabajo es reportar el caso de paciente con rabdomioma asociado a esclerosis tuberosa, tratado quirúrgicamente con éxito.
Caso clínico
Paciente masculino, neonato de 21 días, producto de la primera gesta. Madre de 28 años, que cursó con embarazo normoevolutivo. Se obtuvo mediante operación cesárea por desproporción cefalopélvica, pesando al nacer 4,630 g, talla de 53 cm, Apgar de 6/8. Se detectó soplo cardiaco al nacimiento por lo cual es referido a nuestro servicio.
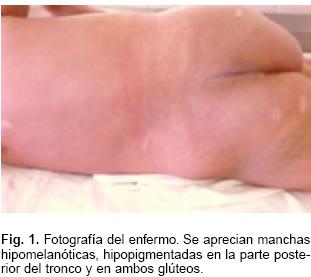
A la exploración física inicial, se encontró con FC: 144X' FR: 34X'. Se observaron múltiples manchas hipomelanóticas, hipopigmentadas en todo el cuerpo (Fig. 1). Con soplo sistólico en mesocardio, sin otras alteraciones. La radiografía de tórax con cardiomegalia grado II. El electrocardiograma en ritmo sinusal, con datos de crecimiento biventricular. Ecocardiograma con múltiples tumoraciones intracardiacas, con masa mayor a nivel del ventrículo derecho, ocupando 2/3 de dicha cavidad, siendo lobulada y condicionando obstrucción parcial del tracto de salida con gradiente de 30 mm Hg (Fig. 2 a). A nivel del ventrículo izquierdo con múltiples tumoraciones pequeñas. Ultrasonido abdominal normal. Tomografía axial computarizada (TAC) de cráneo con calcificaciones subependimarias y a nivel periventricular, sin datos de edema u obstrucción.

Se realizó diagnóstico de rabdomioma cardiaco y esclerosis tuberosa.
Se decidió inicialmente conducta conservadora, por encontrarse el paciente asintomático y sin compromiso hemodinámico. Se hizo seguimiento mediante ecocardiograma seriado, permaneció asintomático hasta los 5 meses de edad, en que desarrolló datos de insuficiencia cardiaca, detectándose por ecocardiografia obstrucción al tracto de salida del ventrículo derecho, con un gradiente Doppler de 60 mm Hg, por lo que se decidió tratamiento quirúrgico (Fig. 2b). Se realizó cirugía con derivación cardiopulmonar de 1 hora y 10 minutos y tiempo de isquemia de 25 minutos. Se resecaron 3 tumoraciones intracavitarias a nivel del septum de salida y de región trabecular del ventrículo derecho de 1.5 cm y de 0.5 cm en su diámetro mayor (Fig. 3).

El estudio histopatológico reportó núcleos deformados con algunos ribetes de citoplasma eosinófilo (células en araña); el tejido se encontró dividido por tabiques fibrosos. Se concluyó rabdomioma (Figs. 4a y 4b).

A 5 meses de la operación el paciente se mantiene asintomático.
Discusión
El rabdomioma cardiaco es la tumoración más frecuente en la edad pediátrica, alcanzando el 45% en autopsias y el 79% series clínicas. Hasta el 75% se encuentran en los menores de 2 años. Es considerado benigno por sus características morfológicas, denominándosele hamartoma debido a que simula estructuras normales en crecimiento desordenado, por lo que no se considera una neoplasia verdadera. Sin embargo, puede presentar manifestaciones muy variadas dependiendo del sitio del tumor, qué estructuras involucre, obstruya o dañe. Puede producir síncope, insuficiencia cardiaca, síndrome de obstrucción caval, hipertensión arterial pulmonar, isquemia pulmonar, cerebral o miocárdica, cor pulmonale, arritmias de cualquier tipo, pudiendo acompañarse en el 15% de Wolf–Parkinson–White, embolia, trombosis y muerte súbita.12 Pueden afectar cualquier parte del corazón, teniendo predilección por los ventrículos. Generalmente suelen ser múltiples, bien circunscritos, lobulados.4,9 Microscópicamente se distingue por la presencia de células anormales típicas, llamadas en "araña", las cuales son grandes, con núcleo excéntrico, conteniendo una masa citoplasmática central, que está suspendida por finos procesos fibrilares irradiados a la periferia.
Nódulos compuestos de miocitos con vacuolas gigantes y grandes cantidades de glicógeno.10,15 El diagnóstico puede hacerse de forma prenatal, a partir del segundo trimestre y generalmente secundario a alteraciones del ritmo, detección del tumor, hydrops, escaso crecimiento fetal y antecedente familiar de esclerosis tuberosa. Postnatalmente suelen detectarse por presencia de soplo, enfermedad valvular obstructiva o búsqueda por diagnóstico de esclerosis tuberosa. En asociación con esta entidad, el espectro clínico es variado, de acuerdo a las estructuras que se encuentren afectadas. Las lesiones hipomelanóticas suelen presentarse hasta en el 97% de los pacientes.6,10,12,16
El síntoma más común son las convulsiones, pudiendo ser refractarias a tratamiento; generar retraso mental severo y desarrollo de hidrocefalia progresiva por las tumoraciones cerebrales. En ausencia de convulsiones el retraso mental es bajo, pero en presencia de retraso mental, las convulsiones siempre estarán presentes.5,9,14
Conclusiones
El rabdomioma cardiaco es un tumor de escasa frecuencia, y de estirpe benigna, que puede ser detectado en forma temprana en la etapa prenatal, que remite en la mayoría de los casos hacia el final trimestre del embarazo. Cuando esto no sucede, debe dársele vigilancia ecocardiográfica ya que se ha demostrado regresión hasta los 4 años de edad. Generalmente no requiere de tratamiento quirúrgico, sin embargo, el tratamiento quirúrgico se recomienda, cuando presenta síntomas o datos que comprometan la vida del paciente pudiéndose evidenciar por arritmias, obstrucción o insuficiencia cardíaca, o ambas, como sucedió en nuestro caso.4,6,9–11,17,18
Referencias
1. Nadas AS, Ellison RC: Cardiac tumors in infancy. Am J Cardiol 1968; 21: 363–366. [ Links ]
2. Lymburner RM: Tumors of the heart: histopathological and clinical study. Can Med Ass J 1934; 30: 368–375. [ Links ]
3. Jansen F, van Nieuwenhuizem O, van Huffelen A: Tuberous sclerosis complex and its founders. J Neurol Neurosurg Psychiatry 2004; 75: 770. [ Links ]
4. Shapiro LM: Cardiac tumours: diagnosis and management. Heart 2001; 85: 218–222. [ Links ]
5. Fesslova V, Villa L, Rizzuti T, Mastrangelo M, Mosca F: Natural history and long–term outcome of cardiac rhabdomyomas detected prenatally. Prenat Diagn 2004; 24: 241–248. [ Links ]
6. Bader R, Chitayat D, Kelly E, Ryan G, Smallhorn J, Toi A: Fetal rhabdomyoma: prenatal diagnosis, clinical outcome and incidence of associated tuberous sclerosis complex. J Pediatr 2003; 143: 620–624. [ Links ]
7. Tworetzky W, McElhinney DB, Margossian R, Moo–Grady AJ, Salle D, Goldmuntz E, et al: Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol 2003; 92: 487–489. [ Links ]
8. Costello JM, Vitullo DA, Yan P, Backer C, Mavroudis C: Left ventricular outflow tract obstruction secondary to a rhabdomyoma. Circulation 2003; 107: 1066–1067. [ Links ]
9. Verhaaren HA, Vanakker O, De Wolf D, Suys B, Francois K, Matthys D: Left ventricular outflow obstruction in rhabdomyoma of infancy: meta–analysis of the literature. J Pediatr 2003; 143: 258–263. [ Links ]
10. Miranda ChI, Muñoz CL, Buendía HA, Aranda FA, Erdmenger OJ, Ramírez MS: Rabdomioma gigante intracardiaco en la etapa neonatal. Reporte de un caso. Arch Cardiol Mex 2004; 74: 49–52. [ Links ]
11. Abad C, Trillo M, Olalla E, Suárez P, Antúnez M, Romero T, et al: Rabdomioma cardíaco y esclerosis tuberosa. Supervivencia tras la resección quirúrgica del tumor cardíaco. Rev Esp Cardiol 1991; 44: 280–282. [ Links ]
12. Lendvay T, Marshall F: The tuberous sclerosis complex and its highly variable manifestations. J Urol 2003; 169: 1635–1642. [ Links ]
13. Osborne J, O'Callaghan F: The management of tuberous sclerosis. Curr Pediatr 2003; 13: 365–370. [ Links ]
14. Serrano BL, Durán PM, Chávez ML, Soriano RJ, Barba RM, Valenzuela EA, et al: El espectro morfológico del complejo esclerosis tuberosa. Análisis de cuatro casos de autopsia. Rev Med Hosp Gen Mex 2000; 63: 12–17. [ Links ]
15. Fenoglio JJ, McAllister HA, Ferrans V: Cardiac rhabdomyoma: a clinicopathologic and electron microscopic study. Am J Cardiol 1976; 38: 241–251. [ Links ]
16. Peña CA, Vasallo PN, Berty PA: Rabdomioma cardiaco biventricular. Reporte de un caso diagnosticado in útero por ecocardiografía. Rev Cubana Cardiol Circ Cardiovas 2001; 15: 52–55. [ Links ]
17. García AA, García TD, Sánchez MF: Rabdomioma de aurícula derecha. Reporte de un caso. Cir Ciruj 2002; 70: 455–458. [ Links ]
18. Beghetti M, Gow R, Hancy I, Mawson J, Williams W, Fredom R: Pediatric primary benign cardiac tumors: a 15 years review. Am Heart J 1997; 134: 1107–1114. [ Links ]

