










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos de cardiología de México
On-line version ISSN 1665-1731Print version ISSN 1405-9940
Arch. Cardiol. Méx. vol.73 n.2 Ciudad de México Apr./Jun. 2003
Editorial
β Fibrilosis ("amiloidosis") sistémicas y cardíacas. Un comentario clínico
Pedro A. Reyes,* Justo Sierra Johnson,** María Elena Soto y Luz Elena Concha**
* Dirección de Investigación. Departamento de Inmunología.
** Pasante en Servicio Social. Instituto Nacional de Cardiología "Ignacio Chávez".
Correspondencia:
Dirección de Investigación. Dpto. de Inmunología.
Instituto Nacional de Cardiología "Ignacio Chávez".
(INCICH, Juan Badiano No. 1, Col. Sección XVI,
Tlalpan 14080 México, D.F.).
Recibido: 13 de marzo de 2003
Aceptado: 31 de marzo de 2003
Palabras clave: Amiloidosis. Miocardiopatía. Diagnóstico.
Key words: Amyloidosis. Cardiomyopathy. Diagnosis. (Arch Cardiol Mex 2003; 73:95-97).
Desde que Rokitansky describió el "hígado lardáceo" y Virchow propuso el término "amiloidosis" para describir una sustancia eosinofílica que creyó era un carbohidrato complejo, celulosa, depositada en la matriz extracelular de diferentes tejidos, esta condición rara ha llamado la atención en Medicina Interna. En la segunda mitad del siglo XX hubo progreso constante en su estudio y se identificó como un material fibrilar con una imagen característica en el microscopio electrónico, afinidad para ciertos colorantes y una conducta específica a la luz polarizada, pero el avance mayor fue la definición precisa del "amiloide" que no es un carbohidrato complejo, como creyó Virchow, sino un estado conformacional de distintas proteínas, que, por razones aún desconocidas, se sintetizan en grandes cantidades por células que han sufrido estímulos por degeneración neoplásica, efecto crónico y sostenido de citocinas, tal vez proinflamatorias o mutaciones puntuales, en genes que codifican las proteínas.
Estos estímulos inician una síntesis proteica con estructura peculiar que genera microfibrillas torcidas sobre su eje, que coalescen y forman placas resistentes a proteólisis, "ahogan" al tejido noble y causan alteración anatómica y disfunción de órganos y tejidos, bien como enfermedad órgano limitada, bien como enfermedad sistémica, en ambos casos irreversible e intratable.
La amiloidosis afecta al corazón, tanto cuando es enfermedad sistémica como en algunas formas órgano limitadas y causa una miocadiopatía restrictiva de muy mal pronóstico y tratamiento complicado y frustrante. Es necesario que el cardiólogo tenga un alto índice de sospecha para orientar el estudio de los pacientes y conocer mejor la enfermedad, de modo que tengamos informes fiables sobre su prevalencia, naturaleza, características clínicas y tal vez podamos coordinar esfuerzos y asignar recursos para la investigación en centros de concentración que faciliten el estudio sistemático de la amiloidosis cardíaca, una enfermedad molecular que tiene interés tanto en su genómica como en la proteómica que le sigue.
La comprensión de estos aspectos, tal vez, dé luz que permita un tratamiento racional de los pacientes que sufren esta enfermedad intratable hasta ahora.
Revisaremos el abordaje diagnóstico
Amiloidosis, (el nombre tradicional persistió a pesar de la propuesta de Glenner, muy razonable, de llamarle β fibrilosis), es un término que designa un grupo de enfermedades sistémicas u órgano limitadas, resultado del depósito tisular de proteínas, muchas y diferentes, que adoptan una conformación molecular esencialmente normal, pero que se sintetiza en exceso y cuantitativamente es anormal. Tal conformación es la de láminas plegadas antiparalelas β, torcidas sobre su eje longitudinal. La conformación es normal en la molécula de proteínas, pero hay diversas proteínas o sus subunidades que adoptan esa conformación en forma anómala y dan origen a fibrillas resistentes a la proteólisis que se depositan en la matriz extracelular. Las fibrillas tienden a agregarse lateralmente, unas a otras y forman "placas" que desplazan y destruyen la arquitectura tisular. Además de las proteínas anómalas en el amiloide, hay proteína P una pentaxina que representa 5% del total de proteínas y pequeñas cantidades de carbohidratos y mucopolisacáridos, pero las proteínas anormales son la base de los tipos de amiloidosis que encontramos en la clínica. Todo "amiloide" comparte estructura y propiedades tintoriales.
En la actualidad tal vez la forma más común de amiloidosis es la asociada a mieloma múltiple (MM). Esa amiloidosis, llamada primaria en clasificaciones antiguas, es debida al depósito de cadenas ligeras de la molécula de inmunoglobulina, que forma el amiloide AL con depósitos en órganos tales como la lengua, la piel y zonas periarticulares con imágenes características, causa también nefropatía y cardiopatía. No se sabe con precisión qué induce la formación de proteína fibrilar en MM; se sabe que hay sustitución de algunos aminoácidos en posiciones específicas en la estructura de la proteína y que tales cambios son más frecuentes en la proteína "amiloide" que en la normal.
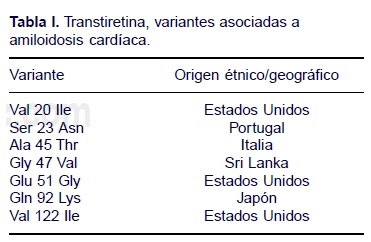
Hoy día se disputan el segundo sitio en frecuencia la amiloidosis secundaria, que resulta de la precipitación, digamos así, de una proteína de fase aguda SAA con generación de amiloide AA. Esa es la amiloidosis de la inflamación crónica, cada vez menos asociada a infección crónica y más a enfermedades como la artritis reumatoide, otras reumáticas generalizadas y algunas neoplasias. El otro competidor, entre las amiloidosis sistémicas, lo representan las formas familiares donde la proteína transtiretina, la vieja prealbúmina, proteína de transporte, se deposita en láminas plegadas cuando existen mutaciones de la proteína que favorecen tal conducta. En estas formas dominan la clínica la neuropatía periférica y la miocardiopatía. Las mutaciones en TTR ligadas al proceso de amiloidosis son poco más o menos 70 a la fecha y causan amiloidosis familiar o esporádica. La primera conocida fue TTRV30M con patrón autosómico dominante en su herencia y asociada a polineuropatía familiar. La TTRV 1221 es la más común entre las que causan miocardiopatía, se observa sobre todo en afroamericanos, consiste en el cambio en el codón 122 de adenina por citosina. Esto ocasiona que en la proteína se sustituya isoleucina por valina y esa enfermedad molecular produce amiloidosis cardíaca. No es la única mutación cardiopática, hay otras más descritas (Tabla I). Hay otras amiloidosis, órgano limitadas, donde participan otras proteínas. Entre ellas están las formas asociadas a diálisis crónica, relacionada con β2 microglobulina, a la enfermedad de Alzheimer y el síndrome de Down con β proteína, y la amiloidosis senil limitada al corazón, así como formas asociadas a cáncer medular de tiroides y a diabetes o insulinoma; de ésas no trataremos más. Como en toda la medicina la historia clínica da pautas a un observador informado. La amiloidosis se sospecha ante síntomas o signos poco específicos como pueden ser: fatiga fácil, pérdida de peso, dolor, púrpura y gingivorragia en un adulto, 99% de las veces mayor de 40 años en quien, al estudiarse, se integran síndromes que reflejan la patología orgánica:
• síndrome nefrótico en una paciente con AR o en un tuberculoso, en una osteomielitis crónica;
• la insuficiencia cardíaca o los trastornos de conducción en un contexto inesperado, e inexplicable,
• la neuropatía periférica y/o autonómica, o neuropatía por compresión, síndrome del túnel del carpo lo más frecuente, también inesperada e inexplicable,
• hepatomegalia o hepatoesplenomegalia,
• síndrome de obstrucción intestinal o enteropatía perdedora de proteína,
• mieloma múltiple con todo su cortejo pleomórfico
Debe mencionarse que algunos signos son sugestivos de esta enfermedad: macroglosia, "hombreras" por depósito peri articular, púrpura peri ocular con aspecto de mapache. Por supuesto, es común la imbricación de más de uno de esos síndromes, pero también puede haber uno dominante, único inclusive.
De allí partimos, ahora tenemos que definir el diagnóstico. Dada la frecuencia de amiloidosis AL, es buena idea hacer una electroforesis de proteínas del suero; se puede encontrar una gamopatía monoclonal, la relación base/altura mayor de 3 y la imagen misma la sugieren, pero 1/5 de los mielomas son mielomas de cadena ligera y no se ven en la electroforesis común, de modo que la prueba de Bence Jones sancionada por el tiempo sobre la proteinura, sigue siendo útil, es casi procedimiento de consultorio. La orina positiva al ácido salicílico, 3 gotas de la solución al 20% por mililitro de orina, precipita, se hace turbia; al calentar a 60 grados se aclara y reprecipita al enfriar. En el laboratorio de hoy se hace inmunofijación en suero y en orina, que identifica las cadenas ligeras. Deben usarse en el escrutinio suero y orina, porque un tercio de los pacientes no tienen inmunofijación positiva en suero. En amiloide AL, 90% de los casos se reconocen con esta estrategia. Pero hay amiloidosis AA y TTR. No hay pruebas de laboratorio simples para reconocer proteína AA, ni formas mutantes de transtiretina.
No queda más que mostrar el depósito tisular de la proteína anormal. Por supuesto, la biopsia de un órgano, el riñón en caso de nefrosis, el corazón si hay miocardiopatía, del hígado si existe hepatomegalia, o del nervio periférico, es capaz de revelar el depósito extracelular, pero esta medida invasiva no está exenta de riesgos y generalmente es innecesaria. La biopsia de médula ósea o la biopsia por aspiración de grasa de la pared abdominal, muy seguras, muestran el depósito de amiloide en cerca de 90% de los casos, de modo que se recomiendan como prueba diagnóstica. De hecho una aspiración de grasa de pared abdominal, que muestra depósitos de amiloide en ausencia de proteína monoclonal, debe hacer sospechar una forma de amiloidosis no relacionada a inmunoglobulinas. Otros sitios de biopsia de rastreo incluyen la mucosa rectal, la encía y la piel; si esto no da resultado, hay que reconsiderar biopsia en otros órganos.
Excepto en la amiloidosis asociada a la fiebre periódica del Mediterráneo, donde la colchicina previene la amiloidosis, no hay aún tratamiento efectivo en amiloidosis. En las formas AL y AA, se busca controlar la enfermedad de base, mieloma, infección, inflamación o neoplasia, con éxito relativo y ocasional; en las formas por TTR no hay nada que funcione.
El pronóstico depende de la topografía de la enfermedad, las formas neuropáticas dominantes o únicas, con frecuencia familiares, tienen el mejor pronóstico para la vida, promedio de sobrevida de 5 años; en las formas cardíacas sólo es de 8 meses. En suma, la amiloidosis es un reto diagnóstico, requiere de un alto índice de sospecha, de uso juicioso de los medios auxiliares de diagnóstico y de comunicación con los diversos especialistas involucrados; con los recursos actuales debe ser diagnosticada sin mayor problema en un hospital. Su patogenia es también un reto para la investigación y probablemente el avance en esos conocimientos nos permita diseñar enfoques de manejo que cambien radicalmente el pronóstico de estas enfermedades.
Referencias
1. Glenner GG: Amyloid deposits and amyloidosis. The β fibrilosis. New Engl J Med 1980;302:1283-92. [ Links ]
2. Gilmore JD, Hawkins PN, Pepys MB: Amyloidosis: a review of recent diagnostic and therapeutic developments. Br J Haematol 1997; 99: 245-56. [ Links ]
3. Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck F, Byxbaum JN: Variant-sequence transthyretin (isoleucine 122) in late onset cardiac amyloidosis in black Americans. New Engl J Med 1997; 336: 466-73. [ Links ]
4. Falk RH, Comnezo RL, Skinner M: The systemic amyloidosis. New Engl J Med 1997; 337: 898-909. [ Links ]
5. Kingman A, Pereira NL: Cardiac amyloidosis. JSC Med Ass 2001; 97: 201-6. [ Links ]

