










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArchivos de cardiología de México
versão On-line ISSN 1665-1731versão impressa ISSN 1405-9940
Arch. Cardiol. Méx. vol.72 no.4 Ciudad de México 2002
Revisión de temas cardiológicos
La meta de la reperfusión en los síndromes isquémicos coronarios agudos con elevación del segmento ST. El gran paradigma: "Lo que hay más allá del flujo TIMI 3 epicárdico: El TIMI 4 miocárdico"
The target of reperfusion in ST - elevation acute coronary syndromes: The major paradigm: "beyond TIMI 3 flow: The TIMI 4 or myocardial tissue - level perfusion".
Eulo Lupi-Herrera,*** Héctor González Pacheco,* Ursulo Juárez Herrera,* Eduardo Chuquiure,* Gerardo Vieyra,* Carlos Martínez Sánchez**
* Médico adscrito.
** Subjefe del Departamento.
*** Jefe del Departamento.
Del Departamento de Urgencias y Unidad Coronaria del Instituto Nacional de Cardiología "Ignacio Chávez" (INCICH, Juan Badiano No. 1, Col. Sección XVI, Tlalpan, 14080. México, D.F.).
Recepción: 30 de abril de 2002
Aceptado: 9 de septiembre de 2002
Resumen
En el tratamiento de los Síndromes Coronarios Agudos con elevación del segmento ST se ha avanzado en la última década de manera favorable en relación a la terapia fibrinolítica (TF), en los procedimientos coronarios intervencionistas (PCI) y con la utilización concomitante de los inhibidores de los receptores plaquetarios IIb/IIIa (IRP). El interés actual en relación al objetivo ha alcanzar en la reperfusión del infarto agudo del miocardio (IAM) ha girado de la arteria responsable del infarto (ARI) a obtener perfusión microvascular-tisular óptima. Se ha puntualizado que el establecer la mejor permeabilidad de la ARI (TIMI 3E) no es sinónimo de que también se ha obtenido en el tejido miocárdico (TIMI 4 M). Sabemos que puede existir disfunción microvascular producto de la microembolización plaquetaria o la ocasionada por la propia reperfusión, misma que esta ligada a los mediadores inflamatorios lo que da origen al "fenómeno de no- flujo", anomalía todas que ocurren en un número no despreciable de enfermos a pesar de haberse obtenido TIMI 3E. Hoy día hay técnicas y tratamientos que van encaminados a identificar y resolver estas anomalías con el fin de mejorar la perfusión microvascular en el IAM. A pesar de existir progresos en las estrategias de reperfusión en el IAM particularmente con el empleo adjunto de IRP y con la TF y que se obtienen en la ARI flujos TIMI 3E en el 50-75% de las veces y con los PCI en el 90-95%, no se han alcanzado reducciones significativas en la mortalidad, mas sí en la frecuencia de la retrombosis de la ARI, de los stents, de reinfartos y en algunos sujetos se observa mejoría de la función ventricular. Por lo tanto, hoy día estamos conscientes de lo que representa obtener perfusión óptima microvascular en el escenario del IAM. El gran paradigma es saber por lo tanto que hay más allá del TIMI 3E y si se alcanzo o no flujo TIMI 4 o miocárdico.
Palabras clave: Infarto agudo del miocardio, reperfusión,TIMI 3E, obstrucción microvascular, fenómeno de no flujo, TIMI 4 M.
Summary
Treatment for ST- elevation acute coronary syndromes (acute myocardial infarction: AMI) has advanced rapidly in the last decade with major improvements in early fibrinolytic therapy (FT), primary percutaneous interventions (PCI) with the aid of platelet glycoprotein IIb/IIIa inhibitors. Recent interest has shifted from infarct related artery (IRA) patency to microvascular perfusion in the evaluation of patients with AMI. It is well known that establishing epicardial patency after AMI (TIMI 3 E) is not synonymus with tissue-level perfusion (TIMI 4M). Microvascular dysfunction due to the roles of platelet and inflammatory mediators in the no-reflow phenomenon occurs in a substancial proportion of patients despite thrombolytic therapy or PCI procedures. Techniques are now available that measure real tissue-level perfusion and also therapy is directed to optimize myocardial perfusion in patients with AMI. Despite advances, contemporary FT strategies with the combination of platelet glycoprotein IIb/IIIa inhibitors restore normal coronary flow (TIMI 3) in the IRA in only 50-75% and PCI achieves TIMI 3 flow rates in 90-95%, but only with modest reductions in mortality, but with significant reductions in rethrombosis of the IRA or stents, reinfarctions and in some patients with benefits in ventricular dysfunction. Therefore moving beyond the importance of TIMI 3 flow, the TIMI 4 flow, or improving tissue-level perfusion in the setting of AMI seems to be the paradigm for the treatment of ST-elevation acute coronary syndromes.
Key words: TIMI 4 M, acute myocardial infarction, reperfusion, TIMI 3 E, microvascular perfusion, the no-flow phenomenon.
Introducción
El espectro clínico y la estratificación
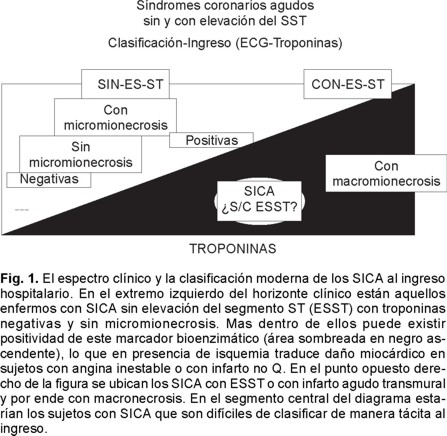
Los enfermos con síndromes isquémicos coronarios agudos (SICA) hoy día se pueden clasificar a su ingreso hospitalario en la clínica, en aquellos que se les identifica elevación del segmento ST (ESST) y los que no tienen este supradesnivel en el ECG de superficie (SEST).1,2 En relación a estos últimos también se estratifican tomando en cuenta el valioso aporte que nos dan los biomarcadores, en especial las Troponinas T e I. Estos biomarcadores anormales reflejan daño miocárdico pero no indican su mecanismo. Por lo tanto una elevación de los mismos en ausencia clínica de isquemia nos debe hacer buscar otras causas de daño cardiaco, por ejemplo miocarditis. Mas en el escenario de los SICA de ser negativas seis a ocho horas después del dolor anginoso, razonablemente se puede decir que no hay necrosis miocárdica, en cambio al ser anormales o positivas enunciar que se trata de un SICA SEST que se acompaña de micromionecrosis (Fig.1).3,4 Lo que correspondería en la práctica a enfermos con angina inestable con daño tisular cardíaco o con IAM no transmural. Más es importante consignar que la estratificación en subgrupos de enfermos con SICA SEST también tiene importancia clínica tomar en consideración: la duración del dolor, la frecuencia e intervalos de los episodios de angina, si éste se desencadena en reposo o con el esfuerzo.5,6 Habrá un grupo que acuden sólo con cambios electrocardiográficos inespecíficos de la onda T o bien específicos en el ECG. Mas hay otros que tienen depresión del segmento ST. En estos a su vez: aquellos con menos y otros con más de 2 mm de infradesnivel del ST los que se les ha conferido el mayor riesgo cardiovascular y que por lo general se acompañan de elevación de Troponinas y de CK-MB. En el extremo opuesto del horizonte clínico de la angina estable están ubicados los enfermos con SICA y elevación del ST y los que por ende tienen troponinas positivas al igual que CK-MB anormales, es decir hallazgo bioenzimático que traduce infarto agudo del miocardio (IAM) clásico o transmural.7,8 Como casi todas las clasificaciones propuestas en medicina tiene ángulos débiles, por ejemplo: hay cierto grupo minoritario de enfermos que a su ingreso nos es difícil de clasificar de manera tácita. Éstos son los que acuden con infartos subendocárdicos laterales, posteriores pequeños, limitados al ventrículo derecho o con bloqueo de rama derecha o izquierda. Mismos que no son identificados a su arribo a los Servicios de Urgencia y si horas más tarde con determinaciones cuantitativas de troponinas, con la utilización de la CK-MB seriada o con el análisis cuidadoso secuencial del ECG o por estudios de ecocardiografía o de medicina nuclear o por el estudio angiográfico coronario, es que podemos llegar al diagnóstico preciso en relación a la magnitud de la afección miocárdica isquémica. Mismos donde nos podemos equivocar involuntariamente y adoptar estrategias terapéuticas contemporáneas que aunque aceptadas no son las más apropiadas para su manejo.9 Sin embargo, a pesar de estas limitaciones ésta parece ser la mejor Clasificación Clínica aceptada hoy día y sugerida al ingreso para los enfermos con SICA (Fig. 1).1-8
Para los SICA con elevación del segmento ST la estrategia terapéutica que ha dado la mayor reducción de la mortalidad en las últimas dos décadas ha sido, sin lugar a duda, la reperfusión exitosa de la arteria responsable del infarto (ARI).10 Sin ignorar el impacto favorable que han tenido también la aspirina, los betabloqueantes, la heparina, los inhibidores de la enzima convertidora de angiotensina y más recientemente las estatinas en la prevención secundaria en la cardiopatía isquémica.11,12 Reciente es el cambio en el pensamiento en el orden terapéutico de además de contemplar la importancia que tiene alcanzar la permeabilidad óptima de la ARI y conservar su flujo, que exista la mejor perfusión en el miocardio y de limitar el daño de la reperfusión.13,14 Por lo que esta revisión se centrará fundamentalmente sobre estos aspectos tan novedosos de la cardiología contemporánea en especial los vinculados a la microembolización y al fenómeno de no flujo postperfusión a nivel del miocardio.
El involucro microvascular en la patología cardíaca
Por estudios de Weber y colaboradores,15 realizados en 1987 sabemos que la vasculatura cardíaca comprende el 35% del volumen del miocardio y que a este nivel tisular hay una respuesta heterogénea al insulto isquémico por existir delicadas diferencias regionales funcionales y metabólicas del miocardio.16,17 Hoy día es posible demostrar variaciones anormales en la perfusión miocárdica utilizando un número importante de técnicas modernas: con la centelleografía miocárdica, con el ecocardiograma de perfusión contrastado y con la tomografía por emisión de positrones por citar algunas.18,19
Aunque existe involucro de la microcirculación en diferentes entidades patológicas que afectan el corazón como son: las cardiopatías congénitas, los procesos metabólicos en los que participa el miocardio, las miocarditis, las cardiomiopatías, la enfermedad hipertensiva sistémica, la hipertensión arterial pulmonar primaria, en la hipertrofia miocárdica, nos centraremos en las conocidas de la cardiopatía isquémica.20
La oclusión de una arteria epicárdica no resuelta da habitualmente como consecuencia síndromes coronarios agudos con elevación del segmento ST o sea infarto transmural. Éste se extiende de la zona subendocárdica al subepicardio de acuerdo a los trabajos clásicos de Reimer y cols. desarrollados en 1977.21 Además de esta variación, existen otras vinculadas a la susceptibilidad a la isquemia relacionadas con el grado de circulación colateral coronaria, misma que a su vez está íntimamente ligada a su desarrollo gradual producto de la isquemia.22 Además habrá que considerar la marcada heterogeneidad en la reserva coronaria, lo que finalmente dará la susceptibilidad individual de los grupos miocíticos al insulto isquémico.23
La vasculatura miocárdica tiene como respuesta reconocida a la isquemia crónica el incremento de vasos (angiogénesis): el aumento numérico de arteriolas, la transformación varicosa y la remodelación de las arteriolas y la de los capilares. Estos cambios estructurales están precedidos y acompañados de disturbios funcionales del endotelio y de los tejidos vasculares de músculo liso dependientes del fenómeno de vasomoción.24-27 Las células endoteliales son tan susceptibles al daño isquémico como son los miocitos, el mismo concepto existe en relación a la injuria post-reperfusión.28,29 Este aspecto se sostiene al quedar consignada la poca capacidad del miocardio de recuperarse al ser reperfundido después de un período largo de isquemia, lo que se conoce como fenómeno de no flujo, descrito por Kloner y cols en 1974.30 Este proceso patológico se sabe que puede iniciarse en las primeras horas de haberse recanalizado la ARI y también que es factible se continúe por varios días. Si bien esto parece una respuesta paradójica a la isquemia, este fenómeno es consecuencia de lesiones miocárdicas que pueden ser profundas o irreversibles, mas tiene su fin que es prevenir el desarrollo de hemorragias en la zona de infarto, o al menos así es considerado.30 Sin embargo esta falta de habilidad para perfundir el tejido infartado es más que una curiosidad patológica, ya que puede comprometer al tejido circunvecino de una perfusión normal en las áreas viables. Estas zonas de flujo coronario limitado o bajo, son las que a pesar del estado de vasodilatación muchos capilares no son competentes, lo que es probablemente debido a que algunas de las arteriolas o de las venas que atraviesan la zona del infarto se vean involucradas.31 A partir de diez minutos de establecida la isquemia existen alteraciones patológicas bien documentadas como son: edema de las células endoteliales, bandas de contracción y de necro sis, cambios estructurales del miocardio que también pueden comprometer la perfusión microvascular.20,30,31
Patofisiología del fenómeno de no flujo
El daño postreperfusión
Verdaderos avances existen de esta causal de la pérdida del flujo normal microcirculatorio coronario, mismas que se han fundamentado en las observaciones experimentales relacionadas con la activación y el acúmulo de neutrófilos y con los ensayos al depletarlos. También con los conocimientos adquiridos acerca de la activación del complemento, de la actividad de las selectinas e integrelinas CD 11b y CD 18 y los estudios concernientes a la generación y liberación de radicales de oxígeno (Fig. 2).32

La activación y la acumulación de los neutrófilos
Inmediatamente después de la recanalización de la ARI ocurre el fenómeno antes citado en el miocardio deteriorado. Sabemos que los neutrófilos son de mayor tamaño que los eritrocitos y menos deformables. Al activarse los primeros se tornan menos moldeables, cambio hemorreológico que favorece el atrapamiento de ellos en los capilares lo que produce taponamiento microvascular.33,34 Se conoce además que los leucocitos tienden ha adherirse al endotelio al igual que las plaquetas cuando el lecho previamente isquémico es reperfundido, hecho que resulta trascendente en tratándose de los SICA ESST. Se ha demostrado que la distribución de los leucocitos en los capilares ha correlacionado bien con la distribución del fenómeno de no - flujo en el animal de experimentación.33 Los investigadores están tratando de identificar cuales son el o los factores que inician de manera temprana el fenómeno de activación- agregación de los neutrófilos.35-37 Hasta ahora este importante fenómeno se ha vinculado al factor activador de las plaquetas que induce la expresión de las integrelinas y la producción de radicales libres (La teoría de los radicales libres de oxígeno).32,38 Al generarse la agregación y la degranulación de las plaquetas se ocasionan decrementos en el flujo coronario cíclico. Un importante estimulante de la quimiotaxis y la adhesión de neutrófilos es el leucotrieno B 4. También se ha notado que la expresión de interleuquina - 6 precede la adhesión de los neutrófilos en el miocardio isquémico.39 Por otro lado, al depletarse los neutrófilos o bloquearlos se asocia a una reducción del tamaño del infarto experimental, lo que sugiere que la acumulación de los mismos favorece el desarrollo de daño miocárdico irreversible. Con la histopatología se ha demostrado que en el animal que es depletado de neutrófilos no aparecen infiltrados de ellos en miocardio infartado y que se atenúa el incremento de la permeabilidad microvascular.40 Hallazgos todos que ponen a los neutrófilos como el principal mediador del daño miocárdico en el binomio patológico isquemia - reperfusión.33-40
El complemento
Existen además datos que han permitido documentar que hay aumento de factores o productos que se derivan del complemento en sujetos con IAM. El tejido miocárdico isquémico genera una proteasa tisular que activa el tercer componente del complemento.41,42 Se han demostrado valores anormales de compuestos del complemento (C3, C4 y C5) de MAC (membrane attack complex) y del complejo macromolecular estable (C5b, C6, C7 y C8).43 La generación de C5a activa los neutrófilos dando como consecuencia incremento en la adhesión célula - célula o célula- substratos, promueve fenómenos de quimiotaxis y la liberación de radicales oxidados y de enzimas proteolíticas.44 Por lo tanto, la activación del sistema del complemento da como consecuencia infiltración de neutrófilos en el miocardio isquémico, amén de la acción de factores quimiotácticos que se han identificados tales como el leucotrieno B4 y otros derivados del ácido araquidónico. Experimentalmente se ha demostrado que depletar o inhibir el sistema del complemento también reduce el daño miocárdico. Este sistema parece jugar un papel importante en el desarrollo de la necrosis miocítica postisquémica al interferir con la integridad de las membranas celulares. La importancia de las moléculas de adhesión de la superficie de los leucocitos en su papel relacionado con la infiltración de los neutrófilos en el tejido miocárdico isquémico ha quedado también validada con ensayos experimentales recientes.45 La extravasación de los neutrófilos en las vénulas postcapilares tiene la siguiente secuencia: agrupamiento de neutrófilos a lo largo del endotelio, activación de los mismos, fortalecimiento de su adhesión, cese de su agrupamiento, taponamiento de ellos y finalmente migración transendotelial. El primer proceso de agrupamiento está favorecido por la interacción célula endotelial-neutrófilo, el que a su vez es mediado por la familia de las selectinas de adhesión [moléculas de glucoproteínas de adhesión] que incluyen: la P selectina, E selectina y L selectina. La P selectina está almacenada en los cuerpos de Weibel-Palade de la célula endotelial y en las plaquetas. Se sabe que tanto la isquemia como la reperfusión producen translocación rápida de la P selectina a la superficie de las células endoteliales de la microvasculatura coronaria.46 La E selectina es estimulada por citoquinas inflamatorias y la L selectina se expresa en la superficie de los neutrófilos. También se conoce que mucho de la adhesión firme de los neutrófilos activados esta mediada por la familia beta dos de las integrelinas y el principal es el ICAM-1 (intercellular adhesion molecule 1). Ésta está compuesta por glucoproteínas heterodiméricas que tienen una subunidad alfa común [CD18] y tres subunidades alfa diferentes (CD11a, CD 11b y CD 11c) exclusiva de los leucocitos. El complejo MAC-1 (CD11b/CD18) es el receptor para CR3, uno de los componentes de degradación del tercer componente del complemento (C3). Se ha encontrado que en sujetos con IAM los niveles de antígeno 1, MAC -1 y de P selectina se encuentran aumentados, lo que a su vez traduce incremento en la función leucocitaria y mayor número de macroagregados microvasculares. Se sabe también que las actividades de adherencia de los neutrófilos está mediada al menos parcialmente por el receptor MAC 1, como es: la adherencia al endotelio y a las plaquetas, a las proteínas de la matriz extracelular, a favorecer la agregación y la quimiotaxis.46,47 La adhesión de los neutrófilos a las plaquetas incluye tanto la P selectina plaquetaria como las integrelinas CD18 neutrofílicas, lo que ejemplifica o solidariza los conceptos modernos de asociación de la trombosis y la inflamación en los SICA. En estudios donde se han utilizado oligosacáridos que bloquean la P y la E selectina se reduce el grado de injuria miocárdica en modelos caninos de isquemia miocárdica y de reperfusión.48,49 Anticuerpos monoclonales bloqueadores de la P y L selectina, del ICAM - 1, del MAC 1 y del CD18 tienen el efecto de abatir la injuria postreperfusión. El mecanismo sería a través de reducir la adhesión, disminuir la liberación de radicales libres y otros oxidantes de los neutrófilos activados previniendo el fenómeno de mionecrosis inducida por oxidantes que resulta del alto contenido de las células inflamatorias que han generado estos productos nocivos.50
Los radicales libres
Además de los mecanismos citados para los neutrófilos y su proceso de taponar los capilares, se sabe que este exceso de células inflamatorias contribuyen a la producción de radicales libres. Los granulocitos pueden producir cantidades substanciales de radicales libres de oxígeno tales como el anión superóxido y radicales hidróxilos, agentes oxidantes enérgicos que dan daño tisular y en las membranas por peroxidación de la cadena de los lípidos.51-54 Hay estudios donde se administra el anión superóxido dismutasa y catalasa dando reducciones del tamaño del infarto. Más aún se ha documentado que estos agentes disminuyen el aturdimiento miocárdico.52-56 Hay observaciones clínicas postprocedimientos coronarios intervencionistas (PCI) que han demostrado incrementos de malondialdehído y de ácido úrico en el seno venoso coronario, lo que sugiere que la reperfusión está asociada a una alta actividad de perioxidación de las membranas y de actividad de la xantino-oxidasa.57,58 Cuando en el IAM se aplica el tratamiento fibrinolítico y se obtiene arteria permeable se encuentran niveles aumentados en la sangre venosa periférica de materiales que reaccionan con al ácido tiobarbitúrico que es un marcador de perioxidación por radicales libres.59 Se han hecho intentos de suprimir la actividad de los radicales libres en la clínica en sujetos con IAM. Algunos investigadores han tratado de conocer la eficacia del flusol, el cual suprime la actividad de los neutrófilos. El estudio piloto de la administración intracoronaria del fluosol en sujetos sometidos a PCI ha demostrado mejoría en la contracción ventricular regional y menor tamaño del infarto.60 Empero estudios clínicos de mayor importancia utilizando el mismo Fluosol, superóxido dismutasa, y prostaciclina no han podido hacer réplica de esos resultados.61-64 Todo parece indicar que hoy día no hay suficiente evidencia de que en enfermos con IAM la terapia supresora de radicales libres bloquee la liberación de éstos o que elimine la función anormal de los neutrófilos en esta parte del escenario clínico.32
Gran interés hay hoy en este rubro, mismo que se enfoca a la disfunción microvascular isquémica y que se observa en el miocardio dañado, pero que tiene la importante característica de ser potencialmente reversible. Zonas alteradas del miocardio que a su vez son las responsables de la falta de respuesta a la terapéutica contemporánea que es utilizada para resolver la oclusión coronaria epicárdica. Esta disfunción microvascular es llamada por algunos autores: de no reflujo capilar, de aturdimiento microvascular, de incompetencia microvascular, de reflujo lento, de flujo bajo o de flujo coronario lento (Fig. 2).65,66 Independientemente de los cambios estructurales y funcionales de las células endoteliales, también pueden ocurrir éstos en el tejido miocárdico normal circunvecino como ha sido comentado previamente.52
La relación de esta disfunción microvascular y el retardo en la normalización de la función contráctil llamada aturdimiento no ha sido completamente dilucidada.32,52 Por otro lado, a la isquemia pre-condicionada que si se le confiere protección en la función contráctil postisquémica, es de aclararse que este mecanismo no protege a la vasculatura del fenómeno de daño de isquemia postreperfusión. La patogenia del aturdimiento microvascular es hoy día investigado profundamente por el enorme potencial terapéutico que se le vislumbra. Se le considera consecuencia de la reperfusión y es diferente de la injuria que resulta de la isquemia prolongada por oclusión coronaria. Es relevante mencionar que el acúmulo de glóbulos rojos y la agregación plaquetaria pueden producir obstrucción microvascular, mas no son la causa dominante o los responsables líderes de la obstrucción microvascular que acontece durante la isquemia. Sólo después de 10 minutos de estado isquémico existe deformidad de la célula endotelial y es en la etapa de reperfusión donde aparece la incompetencia microvascular.20 La activación leucocitaria, la adhesión o la acumulación pueden potencializar tanto el daño miocárdico y la disfunción microvascular postisquémica. Se ha documentado que depletar de los leucocitos circulantes atenúa la lesión postreperfusión y que la inhibición de los factores derivados de su activación previene la disfunción contráctil postisquémica y que favorece el restablecimiento del flujo coronario.32,40
Los tipos de daño postreperfusión
De acuerdo a la autorizada opinión de Kloner RA30,34,52,67 hasta hoy día se han identificado cuatro tipos de daño postreperfusión: a. la injuria letal, b. el daño vascular, c. el miocardio aturdido y c. las arritmias postreperfusión.
La injuria letal postreperfusión
Se le define como muerte del miocito producto de la reperfusión. En esta forma de daño la célula miocárdica que había sufrido daño reversible al final de la isquemia por efecto de la reperfusión sufre cambios letales.68 Es la forma más controvertida de lesión celular por reperfusión tanto en el terreno experimental como en la clínica. Algunos investigadores relacionan el gran daño producto de ésta como otros negando esta posibilidad de muerte celular. 69 Desde el punto de vista experimental es realmente difícil asentar su existencia ya que hay siempre deterioro atribuible a la isquemia como paso previo y sería excepcional obtener modelos experimentales de lesión postreperfusión sin la antesala de la isquemia (Fig. 3). Sin embargo Becker y colaboradores70 con estudios de microscopía electrónica consignan un mayor grado de daño celular irreversible después de la reperfusión que antes de ésta valorado en biopsias. Por lo tanto, este estudio experimental avala este tipo de daño postreperfusión. Sin embargo existen observaciones opuestas como la de Ganz y colaboradores71 utilizando un modelo con dos áreas de isquemia, una reperfundida y otra no, demostrando igual daño tisular en las dos zonas del tejido miocárdico. Se ha tratado de obtener también la respuesta a esta interrogante agregando medicación orientada a reducir el daño con antioxidantes o agentes quelantes, antagonistas del calcio, inhibidores de la enzima convertidora de angiotensina, adenosina o con fluosol.60,61,69,72-74 Los resultados aún son controvertidos y en opinión de Kloner52 es que no existe un consenso en la información experimental actual para poder decir si existe o no el daño postreperfusión de la variedad letal. Desde el punto de vista clínico hay algunas observaciones importantes en este sentido. Por ejemplo, con la administración concomitante de IECA y de trombolíticos en la fase del IAM se ha documentado que se reduce la precarga y la dilatación ventricular.75 Efectos que se pueden explicar de manera más fácilmente a través de las modificaciones en la carga ventricular que a disminución intrínseca en el daño postreperfusión. Bien es conocido el efecto aditivo favorable que tiene la administración de ASA a la terapia fibrinolítica en el IAM y que se traduce en una mayor reducción de la mortalidad que sólo con fibrinólisis.76 Hecho que se ha vinculado principalmente a dar mayor permeabilidad de la ARI que a un beneficio neto en corregir las anomalías de la reperfusión.11,77,78 Todo parece indicar que tampoco desde el punto de vista clínico se puede sostener hasta el momento esta variante postulada de daño postreperfusión. No se debe confundir con la sólida observación que se ha hecho de haberse documentado fenómeno de no - flujo postintervencionismo y que traduce daño microcirculatorio por microembolismo plaquetario aunado al contenido de la matriz de la placa ateroesclerosa embolizado.32

El daño macro y microvascular postreperfusión
En el rubro experimental cuando el corazón se somete a isquemia y a reperfusión no hay duda que aparece daño no sólo en los miocitos sino también en la microvasculatura. Oclusiones coronarias de más de 90 minutos dan como consecuencia al liberarse la obstrucción defectos persistentes de lesión subendocárdica, mismos que se han documentado con marcadores fluorescentes o con carbón negro cuando es inyectado en la vasculatura.30,36,52 La falta de habilidad para reperfundir tejido previamente isquémico es lo que ha designado Kloner como fenómeno de no reflujo.30 Los estudios con microscopía electrónica han demostrado que el daño en la microvasculatura incluye pérdida de la vesículas pinocíticas en el endotelio, vacuolas endoteliales e infiltración neutrofílica. También se ha documentado edema y contractura de los miocitos circunvecinos, lo que a su vez puede contribuir al fenómeno de no flujo.36 Aunque es cierto que algunos de estos daños pueden ocurrir en la vasculatura al final del período de isquemia, también se ha demostrado que éstos se acentúan al acontecer o progresar hacia la fase de reperfusión.30 Ambrosio y colaboradores79 han descrito un aumento de tamaño de la zona de no flujo después de la reperfusión. Con la inyección de microesferas radioactivas se ha demostrado un daño mayor de la perfusión regional postreperfusión, lo mismo se ha observado en relación a la reducción de la reserva coronaria utilizando vasodilatadores dependientes o no del factor relajante del endotelio.34,80-83 Todas estas observaciones llevan al concepto que en efecto ocurre daño vascular por reperfusión en la fase de este proceso. Lo que se ignora es si este mecanismo contribuye directamente o no a la progresión en la muerte del miocito.
En cuanto al panorama clínico hay suficientes datos para aseverar que el fenómeno de no flujo existe en el humano. Lo avalan estudios como el de Schofer y colaboradores84 cuando observan al inyectar marcadores radioactivos intracoronarios en sujetos con IAM defectos perfusorios miocárdicos tanto inmediatamente como a las cuatro semanas de realizada la reperfusión. Más lo que si se desconoce es si el fenómeno de no flujo empeora con el tiempo en el humano.85
Un aspecto interesante a considerar dentro de la injuria postreperfusión de orden vascular es la hemorragia en el seno del área infartada. Esta se ha documentado en los modelos experimentales en el área del infarto agudo tanto con y sin la administración de fibrinolíticos. Se le ha ubicado sólo en el terreno infartado que abarca miocitos con daño irreversible y no parece extenderse a otras áreas, lo que ha hecho pensar a los investigadores que la hemorragia no contribuye al daño isquémico primario de los miocitos y a la muerte celular.86-88 El estudio TIMI [The Thrombolysis in Myocardial Infarction Trial] tiende a confirmar esta observación experimental.89 Los corazones analizados mostraron que las áreas de hemorragia estaban confinadas a las zonas de necrosis y no disecaban hacia los territorios circunvecinos miocárdicos. Por otro lado, estas anomalías hemorrágicas no se relacionaron con las rupturas. Por lo tanto, aunque la hemorragia puede acontecer después de la reperfusión no se puede considerar una variedad aceptada de daño por reperfusión.30,31
El miocardio aturdido
No hay duda que es una forma real funcional de daño por reperfusión y se refiere a disfunción postisquémica ventricular de miocitos viables, donde la isquemia ha desaparecido pero la disfunción miocárdica persiste.90 Es decir, existe una disociación entre el aporte circulatorio y la función contráctil (flujo coronario mayor que la respuesta en la contracción regional ventricular). Bolli y colaboradores91 han demostrado que en el miocardio aturdido este hecho puede estar ligado a la liberación de radicales libres al infundir en un modelo canino de isquemia con duración de 15 minutos seguida de reperfusión el antioxidante MPG (N-(mercaptoproponil) glicina). La infusión de MPG suprime la producción de radicales libres en la reperfusión. Bolli y colaboradores91 con fundamento en esta observación describen al aturdimiento miocárdico, al menos en parte, como una forma de reperfusión mediada por radicales libres. Los megaestudios con fibrinolíticos han demostrado que la recuperación del miocardio isquémico salvado por esta terapéutica requiere de varios días o tal vez de meses.92-94 También en enfermos con angina inestable estudiados con ecocardiografía, ya resuelto el episodio isquémico, se requieren tiempos prolongados para recuperar las anormalidades de la contracción regional ventricular afectadas.95 Post PCI se han visto también anormalidades de la función diastólica que desaparecen 15 - 20 minutos después de haber desinflado el balón.96
Se ha consignado postcirugía de revascularización coronaria, en el ejercicio donde se induce isquemia y después de la revascularización del tejido miocárdico hibernante.97,98 Las implicaciones más importantes que tiene el considerar esta variedad de anomalía postreperfusión son las siguientes: la recuperación de la función no puede descartarse sólo después de que el período de observación ha sido inmediato o en los primeros días, por lo tanto no puede decirse que la terapia de reperfusión (Fibrinólisis o los PCI) han fallado en su cometido de no haberse dado este compás de tiempo. Es importante mencionar que el tejido miocárdico aturdido puede estimularse y contraerse con inotrópicos, por lo tanto en este escenario anormal postreperfusión del miocardio en presencia de insuficiencia cardíaca o de estado de choque se puede y debe utilizar este soporte medicamentoso.99,100 En vista de que el estado de miocardio aturdido puede permanecer por tiempo indefinido o bien aparecer en el momento de realizarse el ejercicio, es importante en el enfermo isquémico convaleciente estar alerta o consciente que para autorizarse el esfuerzo, es menester cerciorarse por medio de los estudios que analizan de la función ventricular (ECO - estudios de perfusión miocárdica nuclear) que esta condición anómala miocárdica no está presente antes de que se le permita al enfermo efectuar este tipo de actividad física.
Las arritmias postreperfusión
Se pueden definir como aquellas que ocurren segundos después de haberse restaurado el flujo coronario tras el episodio de IAM. De hecho son fáciles de desencadenar experimentalmente en modelos animales tras períodos breves de cinco a diez minutos de oclusión coronaria y de liberarse la obstrucción del vaso epicárdico. Sin embargo si el período de isquemia se prolonga por una o tres horas y es seguido de reperfusión, estas arritmias malignas también aparecen, pero sin incrementarse significativamente. No hay duda que éstas ocurren en enfermos con IAM, pero existe la interrogante muy razonable que sean originadas por efecto directo de la reperfusión.101 Las razones que permiten cuestionar su origen directo de la propia reperfusión en el humano son las siguientes: a. se pueden incluir sin tener una demostración fehaciente de que existió prueba angiográfica de reperfusión de la ARI, b. las arritmias que ocurrieron antes de la reperfusión se pueden llegar a considerar como tales y c. las arritmias que acontecieron días o semanas después de la reperfusión se pueden abarcar sin tomar en consideración la posibilidad de reoclusión coronaria. Este tipo de arritmias se ha documentado en promedio en el seis por ciento (rango de cero a 17%) de los sujetos trombolizados.102 Lo que sí es claro es que su frecuencia es mayor cuando ha pasado un intervalo corto entre la oclusión coronaria y la administración de la terapia fibrinolítica, tal como se ha observado en los modelos experimentales. En general, los estudios más importantes donde se ha empleado la terapia fibrinolítica no han demostrado un incremento claro de estas arritmias malignas que ponen en peligro la vida del enfermo y que puedan ser atribuidas a la reperfusión.103 Hay estudios con o sin tratamiento de reperfusión en donde se ha visto durante el transporte del enfermo al nosocomio que la frecuencia de estas arritmias es similar en las dos condiciones de conducta terapéutica. La observación en estudios como el ISIS-2 y el GISSI sobre la reducción de fibrilación ventricular en sujetos trombolizados en el período de hospitalización da evidencia de que la reperfusión no tiene una influencia negativa en generar arritmias malignas ventriculares.104,105
Todo parece indicar que la verdadera incidencia de las arritmias ventriculares postreperfusión es realmente baja. Tampoco predicen que documentarlas traduzca éxito de la reperfusión de la ARI. La mejor explicación de su presencia la da el hecho de que en la clínica la mayoría de los enfermos son llevados a reperfusión con una ventana de cuatro a seis horas con áreas donde ya se ha consumado el daño miocárdico. Por lo tanto, las arritmias presumiblemente atribuidas a la reperfusión son principalmente producto de la isquemia miocárdica y del propio infarto en evolución. Se han investigado otras posibles causas como es el efecto de la liberación de radicales libres. Empero no se ha logrado demostrar que con la administración de superóxido dismutasa se reduzca la frecuencia de taquicardia ventricular en los primeros 15 minutos de haberse obtenido la permeabilidad de la ARI y por lo tanto en la fase de la reperfusión miocárdica. Lo que sí es importante consignar es que éstas sí pueden ocurrir en individuos con cardiopatía isquémica, dar muerte súbita tras períodos breves de déficit irrigatorio miocárdico o de oclusión coronaria, post liberación de la obstrucción o por isquemia inducida por el ejercicio. Más lo relevante a dejar claramente asentado es que no debe existir el temor de inducir ocasionalmente estas arritmias, que son muy raras por efecto directo de la reperfusión en el escenario del IAM cuando se emplee cualquiera de los métodos de reperfusión modernos que son los instrumentos capaces de disminuir la morbimortalidad del IAM.101-105
La importancia de la embolización en la cardiopatía isquémica aguda
Existen trabajos de la última década que han marcado la trascendencia que tiene la microembolización en esta patología en sus etapas de agudización.106-110 Mas sólo es reciente la posibilidad de documentarla en la clínica, gracias a los avances tecnológicos modernos, particularmente los vinculados a la imagenología cardiovascular. Lo que incluye: la resonancia magnética nuclear, la ecocardiografía miocárdica contrastada, y también con los novedosos estudios de medicina nuclear. Mediante esta tecnología en una proporción significativa o no despreciable de enfermos se ha documentado obstrucción microvascular. Más aún lo más trascendente desde el punto de vista clínico es que esta micropatología vascular se ha ligado a evolución no favorable e incremento en la mortalidad. Ligada a esta valiosa observación también se han buscado nuevos caminos terapéuticos para contrarrestarla y con ellos se ha demostrado que es posible abatir la morbilidad y posiblemente la mortalidad que resulta de la microembolización en algunos de los escenarios agudos de la cardiopatía isquémica.
Lo más trascendente es que nos obliga a abrir la mente en aspectos terapéuticos que antes no se consideraban relevantes en la clínica. Tal es el caso al referirnos a los procedimientos más utilizados hoy día para resolver la problemática de arterias epicárdicas ocluidas en el infarto agudo del miocardio o parcialmente obliteradas como en la angina inestable. Si bien la primera meta de la reperfusión en el IAM ha sido obtener flujo TIMI - 3 epicárdico, ésta no parece ser hoy día el solo punto terapéutico final deseado.111 Ya que lo que es óptimo es alcanzar también una perfusión normal a nivel miocárdico y que incluye como premisa la permeabilidad completa de la microcirculación, para entonces decir que se alcanzó flujo TIMI - 4 o miocárdico. Ésta es una de las razones por la que con los métodos modernos de reperfusión no es posible reducir la mortalidad del IAM a cifras cercanas al cero. Con los comentarios hechos previamente también queda claro que hay otros aspectos que están vinculados a la propia reperfusión: el daño ocasionado por ésta, el fenómeno de no flujo, el miocardio aturdido, el hibernante y aspectos aún desconocidos como es la consecuencia de liberar la propia obstrucción microvascular. Patologías isquémicas que tienen un punto inicial de partida: la placa aterosclerosa vulnerable que sufre erosión o fractura (Fig. 4). Situación que deja expuesta la matriz subendotelial y en donde su principal contenido de masa oclusiva es la plaqueta [trombo blanco], mas hay trombosis también donde predominan los glóbulos rojos, con oclusión completa o subtotal en un proceso no estático en sus componentes y que a su vez da origen a los síndromes coronarios agudos de infarto con o sin elevación del segmento ST.109 Es así como en su evolución se ha identificado el fenómeno de la microembolización con alojamiento de estos fragmentos plaquetarios y del contenido de la matriz en los microvasos coronarios distales, proceso que antes se consideraba como una condición no frecuente. Este mecanismo se ha vinculado como una de las posibles causas por la que se generan arritmias y es causa de episodios de muerte súbita.107,108 Es de llamar la atención que no hay muchos estudios importantes donde el área histológica relacionada con el infarto se ha investigado sistemáticamente en el post mortem de estos sujetos. Aspecto que se puede considerar como una deficiencia en nuestros conocimientos actuales, particularmente cuando la terapia trombolítica o la de reperfusión de la ARI con catéteres son nuestros principales métodos terapéuticos para la atención del IAM y ambos son capaces de promover la embolización (Fig. 5). Los PCI se iniciaron hace más de dos décadas y durante todo este tiempo hemos subestimado la posibilidad de que esta situación patológica o bien se le ha considerado muy rara o sólo era conferida al terreno de los injertos de safena añosos y degenerados cuando éstos eran manipulados con catéteres.112 Porqué no nos hemos hecho la pregunta antes: ¿qué sucede con el material de la placa ateromatosa al ser instrumentada? En realidad es hasta hace poco que la opinión de lo raro o lo frecuente de la posibilidad de embolización a la microcirculación ha sido posible contestarla y poder decir si en realidad esta situación derivada del manejo medicamentoso farmacológico o del intervencionismo es trascendente, o bien es irrelevante.


La patofisiología moderna
El significado de la microembolización
El concepto de la microembolización no es realmente nuevo o al menos el de la participación de la microvasculatura en la circulación coronaria. El-Maraghi y Genton E113 describen en un artículo que se debe considerar clásico, la frecuencia de estas anomalías en un grupo de enfermos con diferentes patologías cardiovasculares y hacen patente la relevancia de la tromboembolia de la fibrina y del material plaquetario y su relación con la muerte súbita. Encontraron involucro de daño microvascular en 19/162 enfermos con cardiopatía isquémica y en seis de 47 sujetos sometidos a cirugía cardíaca, describiendo trombosis microvascular, miocitólisis focal y lesiones miofibrilares. En ellos destaca la importancia de estas alteraciones patológicas en la cardiopatía isquémica y es de hacerse notar que lo hacen tanto para el ventrículo izquierdo como para el derecho. Es por estas mismas fechas cuando a partir de los trabajos de Folts y colaboradores y de Willerson y coautores quienes utilizaron un modelo experimental de injuria endotelial coronaria, se empieza a destacar la importancia de las microembolias, particularmente las de origen plaquetario.114-117 Con el pasar del tiempo la patofisiología de la microembolización coronaria se ha puntualizado y se puede consignar de la siguiente manera (Fig. 6) A partir de una placa ateroesclerosa vulnerable que por lo general dará menos del 60% de obstrucción del lumen coronario, puede perder su estabilidad y entrar en actividad.118 Misma que en esta época moderna pudiera eventualmente identificarse en la clínica por estudios de resonancia magnética nuclear.119 Sin embargo, por lo general la actividad y la fractura de las mismas se traduce en un evento clínico (SICA con o sin elevación del segmento ST). A partir de la erosión o la ruptura de la misma da como consecuencia obstrucción completa (trombo rojo, con predominio de glóbulos rojos sobre las plaquetas) por períodos de 6 - 8 horas en el 80% de los enfermos y ocasiona IAM o bien da una trombosis suboclusiva (trombo blanco: rico en plaquetas) ocasionando los síndromes de angina inestable o de infarto no Q. Mismos en los que es relevante consignar que también pueden tener períodos transitorios de oclusión coronaria total que se han documentado del 20 - 40% de los enfermos y que duran habitualmente menos de una hora.119,120 Recientemente Lindhal y colaboradores han realizado un estudio muy importante en el cual correlacionan la determinación cuantitativa de troponina I, la magnitud de lesiones coronarias significativas y la presencia de trombos.5 Los autores consignan que las lesiones coronarias no significativas son rara vez observadas o están ausentes con troponinas positivas. Lo frecuente es documentar obstrucciones bi o trivasculares importantes y lo más trascendente es observar trombosis de las arterias coronarias alrededor del 30% dentro de la primera semana y con mayor incidencia a medida que avanzan los días. Substrato que sugiere o da mayor posibilidad de microembolización repetida durante este período de actividad de la placa aterosclerosa en los SICA sin elevación del ST. Lo importante al tema es que ambas lesiones pueden ser embolígenas a la microcirculación coronaria. Máxime que se sabe que todo este proceso no es explosivo - estático, por el contrario es agudo y muy dinámico, en donde el binomio trombosis - fibrinólisis endógena es lo que predomina dando grados diferentes de obstrucción epicárdica coronaria que se traducen en el ámbito experimental en variaciones cíclicas del flujo coronario y que tienen en la clínica la expresión de episodios de angor de duración variable y habitualmente con alteraciones en el ECG de superficie. La patofisiología de la obstrucción microvascular (OMV) tiene su origen en este primer escenario a partir de la obstrucción total o subtotal de la arteria epicárdica responsable del infarto. Se ha validado que estos fenómenos también pueden ser producto de la manipulación endovascular por los PCI tales como: la angioplastía con balón, la colocación de mallas endovasculares (stents) y la rotaablación endocoronaria, pero también recordemos que puede resultar por algo más común, la acción fibrinolítica exógena (la terapia trombolítica) (Figs. 5 y 6).121,122 Se ha demostrado que en este escenario clínico tan dinámico en donde se deja trombina libre se incrementan en la circulación coronaria en cuestión de segundos o minutos productos tales como el ADP, la serotonina, el tromboxano A2 o su metabolito el tromboxano B. Todos ellos son capaces de favorecer u ocasionar vasoespasmo y los fenómenos de adhesión - agregación plaquetaria dando origen a la formación de microtrombos ricos en receptores de glucoproteínas IIb/IIIa, lo que confirma la importancia de esta estructura tromboembolígena en la génesis de lo que finalmente da focos de micromionecrosis. Es una alteración histológica que se ha documentado hasta en el 40-50% de los materiales estudiados. Es de mencionarse también que junto con el material plaquetario es posible que se acompañe del contenido de partículas la matriz de la placa ateromatosa rota. Existe incremento de las células endoteliales en la sangre periférica de sujetos con SICA al ser comparados con aquellos que tienen SIC crónicos o sólo angina de esfuerzo. En modelos experimentales Eguchi y colaboradores123 han demostrado la pérdida de la integridad a la adhesión plaquetaria, además de lo comentado referente a las plaquetas y los leucocitos. Por lo tanto, es factible que los constituyentes de la placa, de la matriz y las células endoteliales puedan ser embolizadas. Independientemente del daño descrito con antelación, el efecto en la clínica o el panorama parece poder resumirse: en presencia de microinfartos y desarrollo de arritmias potencialmente malignas capaces de dar muerte súbita.108,113 Además hay reducción de la reserva coronaria, disfunción zonal de la masa ventricular o global y caída en disfunción ventricular con mayor morbimortalidad, como se ha demostrado en algunos seguimientos clínicos donde se ha investigado como causal la pérdida de la permeabilidad de la microcirculación coronaria (Fig. 7). Sabemos que tener una arteria epicárdica obstruida o flujo TIMI epicárdico 0 representa un escenario de necrosis absoluta, excepto en aquellos enfermos que desarrollan circulación coronaria ipsi o contralateral. El tener flujo TIMI 2E se consideraba en el pasado como un resultado terapéutico útil, mas hoy día equivale a una mortalidad similar a la del TIMI 0 - 1 (Figs. 8 y 9).111 Existen dos posibles explicaciones para que exista TIMI 2E inmediatamente post reperfusión en el IAM. Puede ser secundario a la presencia de trombo residual, estenosis significativa remanente o disección de la arteria coronaria lo que se traduce angiográficamente en arteria permeable con flujo del contraste reducido en ella y distal en el miocardio. Causas que fácilmente se pueden identificar en la arteria epicárdica por medio de la arteriografía coronaria. En esta época moderna de la aplicación de mallas endocoronarias difícil es de aceptar que el flujo TIMI 2E sea consecuencia de estenosis residual de la arteria epicárdica. Por lo tanto, la razón más importante parece ser la obstrucción microvascular que impide el vaciamiento del flujo epicárdico por los dos mecanismos genéricos: a. microembolismo plaquetario y acúmulo de neutrófilos o b. por daño post reperfusión: donde aparece agregación de neutrófilos, liberación de radicales libres, disfunción endotelial y espasmo microvascular. En enfermos con TIMI 2E por diferentes técnicas y en especial con la ecocardiografía miocárdica contrastada con microburbujas sonicadas de perfluorocarbono se ha demostrado que prácticamente en la totalidad de ellos no existe flujo microcirculatorio.84,85 También es del conocimiento actual que con este mismo método con TIMI 3E por lo menos del 20 al 40% no existe flujo TIMI miocárdico normal o lo que se designaría como TIMI 4 lo que se han vinculado a mayor morbimortalidad en la clínica.




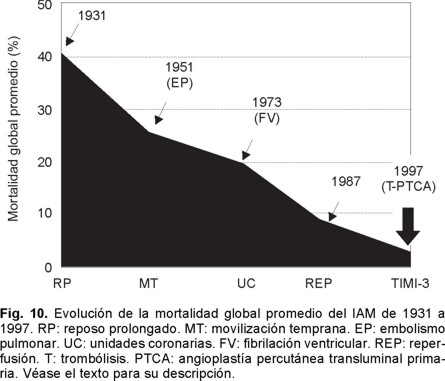
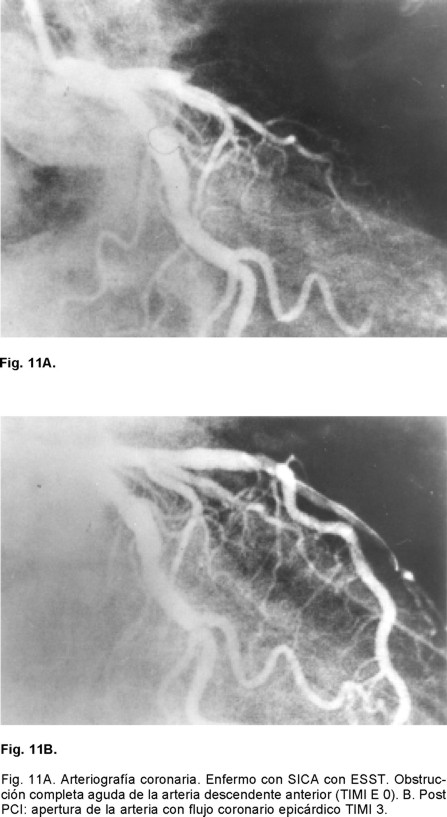
Sabemos que hace 70 años la mortalidad del IAM era cercana al 40%, misma que se logró reducir al 25% a principios de 1951 con la simple maniobra de la movilización temprana y al 20% con la creación de las Unidades Coronarias en 1973. Es en 1987, con la introducción de la terapia fibrinolítica que se establece un nuevo descenso para considerarse cercana al 8%. Ya perfeccionada la terapia de reperfusión y con la introducción de los PCI (angioplastía con balón y mallas endovasculares coronarias) es que tenemos un resultado que oscila del 4 al 6% de mortalidad del IAM (Fig. 10). Es por eso que hoy día se acepta que esta modalidad terapéutica es la que ha sido capaz de brindar el más importante descenso de la mortalidad en el rubro del IAM transmural. Entre los factores más importantes de los que depende alcanzar esta reducción está el factor tiempo relacionado al momento en el que se de el tratamiento o ventana terapéutica en la que se aplique la terapia de reperfusión. La magnitud de miocardio salvado determinada experimentalmente para el ventrículo izquierdo es cercano al 70% si la reperfusión se realiza en la primera hora a partir de haberse ocluido la arteria epicárdica. Disminuye al 40% a la tercera y al 20% a la sexta, para ser menor del 5% a las 24 horas. Para el ventrículo derecho Lester y colaboradores124 han encontrado que el área de infarto a la hora de reperfusión es de tan sólo un cinco por ciento, a las cuatro horas es de un 10% y a las ocho es del 20%. Por lo tanto, el acortar el tiempo de la reperfusión para cualquiera de ambas cámaras ventriculares sería igual a menor daño miocárdico o mayor magnitud de miocardio salvado. Esta es una de las razones por la que después de seis horas el beneficio de la reperfusión de la arteria epicárdica decae significativamente para considerarla casi nula después de doce horas. Lo que se aplica tanto para el tratamiento fibrinolítico como para los PCI. Si en la ARI no se obtiene permeabilidad inmediata la mortalidad es del 17.5%, lo que equivale a remontarnos a la consignada en los años de 1970. Si la ARI está permeable a los 90 minutos pero se le encuentra ocluida al séptimo día, la mortalidad es del 12% lo cual nos ubica en la mortalidad documentada a principios de 1980. Si la ARI está permeable a los 90 minutos y al séptimo día la mortalidad es del 4.7%. Por otro lado, la importancia de la calidad del flujo epicárdico post reperfusión resulta extraordinariamente relevante. Las primeras observaciones en este sentido con la terapia de reperfusión lítica nos demostraron que cuando el flujo epicárdico es TIMI: 0 -1 la mortalidad del IAM es del 8.9%, al ser grado TIMI: 2 la mortalidad es del 7.4% y con TIMI: 3 epicárdico es del 4.4% (Figs. 8 y 11 A - 11 B). Pero por fortuna hemos avanzado en el conocimiento en este sentido y con la valiosa información del estudio GUSTO II b (The Global Use of Strategies to open Occluded Coronary Arteries in Acute Coronary Syndromes) sabemos que con ACTP de obtenerse flujo TIMI 0-1-2 la mortalidad es del 18 - 20%, cifras que resultan casi del doble de las documentadas con trombólisis para los mismos flujos, hecho interesante que nunca se ha explicado claramente.125 En cambio de obtenerse flujo TIMI 3E la mortalidad a treinta días es de tan sólo 1.6%, observándose menor tamaño del área infartada y mayor índice de miocardio salvado. Lo que establece sólo una diferencia del 2.8% entre las dos estrategias de reperfusión cuando se alcanzan flujos TIMI 3E (Fig. 8). Para llegar a estas conclusiones, frente a los enfermos es menester tomar en consideración los datos relevantes proporcionados por Gibson y colaboradores en relación al análisis angiográfico de la perfusión miocárdica.126 Mismo que se ha valorado por dos métodos pero que al final sólo traducen que existe perfusión a nivel de la microvasculatura. Empero hay que recordar que existen varios factores de los que también depende el flujo TIMI epicárdico como son los de orden: hemodinámico, la anatomía local de la arteria, la propia técnica del procedimiento, el trombo intracoronario y aspectos derivados del intervencionismo: la estenosis residual, la disección involuntaria de la arteria coronaria y menos relevantes parecen la aplicación de las mallas endocoronarias (Fig. 12).126 Excluidas estas causales, quedarían las dos más importantes a considerar: la obstrucción microvascular y las que son producto del fenómeno de no flujo en la microcirculación. El flujo TIMI en la microcirculación se expresa o se visualiza en la angiografía coronaria como áreas de condensación de aspecto esmerilado muy finas del material de contraste a nivel del miocardio. Esto es lo que los autores anglosajones designan en su lengua como "blush" y que correspondería a "TIMI miocárdico 4".127-130 Si bien las anomalías de la microcirculación se han demostrado de manera fehaciente por los diferentes métodos citados con antelación, si se cuestiona si lo que documentamos es también producto de edema miocárdico y/o del daño por reperfusión. Sin embargo este patrón de alteración microcirculatoria ocurre tempranamente (en las primeras horas o días), lo que hace menos probable que sea por daño por reperfusión. Es más, se le ha documentado en sujetos que van a PCI (rotaablación coronaria) y tras la práctica de ellos aparecen cambios miocárdicos en los estudios de perfusión nuclear, lo que sugiere embolización a la microcirculación (Fig. 13). 121,122 Con la utilización de catéteres con redes o sistemas de aspiración se han recuperado fragmentos del material que hubiesen sido embolizados. Por lo tanto, en la clínica se puede decir que este mecanismo se sugiere como la causa primordial, al menos en etapas tempranas, de las anomalías documentadas en la microcirculación. Mas es innegable que en el caso de IAM transmural reperfundido deberán estar presentes los dos mecanismos, el inicial ligado a la microembolización y el tardío aunado a los fenómenos de la injuria post reperfusión (Fig. 5). Lo importante es que la obstrucción microvascular está vinculada a mal pronóstico aún de obtenerse la permeabilidad deseada de la arteria epicárdica. Wu y colaboradores131 han demostrado que a 25 meses el 90% de los enfermos sin OMV están libres de muerte-reinfarto e insuficiencia cardiaca. De existir OMV tan sólo el 55% lo estarán. Es más, con estudios de resonancia magnética nuclear se ha podido catalogar la magnitud de los defectos en áreas grandes, moderadas y pequeñas de OMV con diferencias estadísticas en morbimortalidad de cada una de estos grupos y directamente proporcionales a la magnitud del defecto documentado en la resonancia magnética nuclear. Uno de los primeros estudios en llamar la atención en la posibilidad de microembolización ligada a PCI fue el CAVEAT (Coronary Angioplasty Versus Excisional Atherectomy) donde la incidencia de IAM fue superior a la esperada. Se encontró una frecuencia de IAM del 3% para ACPT y del 6% para aterectomía rotacional. En este estudio prospectivo la elevación de la CK-MB a cifras tres veces por arriba del valor basal fue de 8% para la ACTP y del 19% para la aterectomía.121 En el estudio CAVEAT II sobre intervenciones en libramientos de venas safenas la incidencia de IAM fue considerablemente mayor 15 y 24% respectivamente.122 En nuestro medio Vallejo E y colaboradores132 revisaron 204 angioplastías electivas y 62 primarias. El fenómeno de no flujo predominó en hombres, con edad promedio de 56 años y fue la diabetes mellitus un factor de mayor riesgo para observarlo. La incidencia global del problema fue del 5.2%, 16.1% para la angioplastía primaria y del 1.9% en los procedimientos intervencionistas electivos. Por lo tanto, los autores no la consideran como una complicación rara o infrecuente particularmente en el escenario del IAM. Este importante hallazgo de todos los estudios previamente mencionados dio origen a ciertos debates, si los procedimientos intervencionistas utilizados creaban fuga enzimática, eran capaces de dar seudoinfartos o microinfartos.66 En el seguimiento a largo plazo se demostró un exceso de mortalidad para la aterectomía y aquellos que fallecieron presentaron durante el procedimiento cambios dinámicos del segmento ST muy sugestivos de IAM no transmural o subendocárdico. Varios estudios posteriores han demostrado que a mayor elevación de la CK-MB post PCI hay mayor probabilidad de muerte.121,122 Así cuando la elevación de CK-MB es de cinco veces el valor basal, la mortalidad al año es de 6 -7%, de ser 10 veces el incremento de CK-MB es del 13%. La posible causa de este hallazgo es la microembolización coronaria ya que otras eventualidades son menos frecuentes o resultan menos probables como factores causales tales como el tiempo de isquemia ocasionado por el balón que es habitualmente demasiado corto para ocasionar necrosis per se. Las oclusiones abruptas de ramas coronarias epicárdicas hoy día son poco frecuentes (< 3%) y la oclusión sú bita coronaria transitoria se le ve en menos del 1%.135 La incidencia por lo tanto de grados diversos de micromionecrosis, consecuencia de la embolización coronaria, al menos significativa no parece despreciable. Valorada utilizando biomarcadores como la Troponina T o I parece cercana al 30 - 40%. Si bien la identificación alta de troponinas post intervención parece importante, también lo es cuando se documentan elevaciones de la misma antes de realizar los PCI.136,137 Así Hamm y cols138 han demostrado que hay una respuesta diferente al tratamiento con bloqueadores de los receptores plaquetarios. Aquellos con Troponina T positiva al ingreso responden favorablemente al inhibidor del receptor plaquetario por el bloqueo del mecanismo citado a nivel de la microcirculación, además de reducir la frecuencia del trombo en la arteria epicárdica sometida a ACTP o a la colocación del stent.


Parece existir cierto perfil del enfermo prono a desarrollar con mayor frecuencia microembolización. Los estudios demográficos señalan que la Diabetes Mellitus es un factor prominente de riesgo, lo que puede deberse a una enfermedad más difusa coronaria o tal vez a que el enfermo diabético ya haya tenido episodios subclínicos de microembolización y por lo tanto menor capacidad de adaptación a eventos subsecuentes de microembolización, o en relación a la inflamación o tal vez vinculada con la insulina o a otros factores metabólicos. Esta población de diabéticos es la que parece tener más riesgo a la microembolización pero también es en la que más beneficio se ha demostrado con el empleo de bloqueadores de los receptores plaquetarios IIb/ IIIa cuando estos enfermos son enviados a procedimientos de revascularización coronaria percutánea. Tal vez también estén más propensos aquellos sujetos con placas aterosclerosas friables lo que se traduce por elevaciones de la proteína C reactiva, del factor de necrosis tumoral, de las interleucinas o por moléculas de adhesión u otros marcadores de inflamación que se han vinculado de manera reciente al pronóstico a largo plazo en la cardiopatía isquémica aguda.139,140
El escenario clínico y la microembolización
En la angina inestable y en el infarto no Q la disponibilidad de contar con biomarcadores más sensibles que la CK-MB como son las Troponinas T e I, permiten pensar en la posibilidad clínica de microembolización y que son enfermos donde ha ocurrido micromionecrosis (Fig.1). Sabemos que, en general, los biomarcadores son más sensibles y específicos y menos onerosos que la imagenología para el diagnóstico de necrosis. Para que con la ecocardiografía se detecten anormalidades segmentarias de la pared ventricular debe haber por lo menos un 20% de daño y más de 10 gramos de tejido cardíaco lesionado para que sea detectado por un estudio de medicina nuclear perfusorio.2 Definitivamente la positividad de las Troponinas en estos SICA indica una mayor mortalidad al ingreso y a los treinta días de seguimiento y es muy superior a la que se consignaba en el pasado (no mayor de cuatro por ciento).3,5,8,141 Se sabe que de acuerdo a la escala clínica TIMI de riesgo para mortalidad, ésta es proporcional al número de factores de riesgo coronario y la letalidad puede llegar a ser tan alta como 15-16% a treinta días.7 Más aún la mortalidad de este subgrupo de enfermos con SICA, que cada vez es más frecuentemente visto en los Servicios de Urgencia en relación al infarto Q, no se ha demostrado que se haya modificado desde 1975 hasta el año de 1997 (período de 22 años). A diferencia de lo que ha sucedido con el infarto Q donde la mortalidad se ha logrado disminuir de 24% a 14% en el mismo período y que por otros megaestudios con reperfusión lítica e inhibidores de los receptores plaquetarios IIb/IIIa (IRP IIb/IIIa) en forma global es cercana al 6%.142 Los valores de troponinas negativos nos indican que la mortalidad para este grupo de SICA SEST es menor al 3%, empero que ésta guarda un ascenso hasta del 12% con troponinas elevadas con un comportamiento en paralelo con la posibilidad de que ocurra reinfarto. Sin embargo, se ha demostrado que existe un techo o límite de elevación para las Troponinas donde la posibilidad de muerte o de reinfarto es similar a la observada para el grupo de bajo riego cuando este marcador biológico es negativo. Lo que sugiere que esta población puede corresponder a enfermos con infartos transmurales consumados no indentificados o con zonas dispersas de islotes de necrosis miocárdica, donde la posibilidad de reinfarto ya no es factible o es muy remota.5,6 Es importante mencionar que si bien existe un espectro de la relación beneficio utilización temprana de PCI y valores de troponinas en estos enfermos, donde el extremo está caracterizado por cifras de troponinas elevadas (con valores cercanos a los que se observan en macronecrosis) es éste el grupo donde la utilización de IRP IIb/IIIa no parece dar el impacto favorable deseado o el esperado en la microcirculación, sin perder el beneficio demostrado en megaestudios sobre la arteria epicárdica a ser instrumentada. De hecho, se sabe hoy día de acuerdo a un metaanálisis muy reciente, que incluye los principales estudios con el empleo de IRP II b/III a, en el cual se analiza el impacto de esta modalidad terapéutica en este escenario en particular, que son capaces de reducir la mortalidad absoluta en uno por ciento, e independientemente si los enfermos son o no enviados a procedimientos de revascularización coronaria temprana y si tienen o no troponinas positivas. Mas los autores consignan que el grupo que más beneficio devenga con esta modalidad terapéutica es aquel donde los biomarcadores citados son anormales. Ambas observaciones sugieren que éstos dan efecto benéfico impidiendo la microembolización.143,144 Mas recordemos de la fisiopatología que éste no es el único mecanismo por el cual se afecta la microcirculación y que la terapéutica futura debe de encaminarse hacia otros caminos que ya se han identificado como es el proceso de la inflamación (aumento de Proteína C reactiva), aspecto que deberá ser validado en estudios clínicos del futuro (Figs. 2 y 6).139,140
El infarto agudo del miocardio
Un problema mayor en el tratamiento del IAM es la potencialización de la masa de fibrina libre, que es eventualmente embolígena, promovida y favorecida tanto por la propia fibrinólisis endógena como la exógena (terapia trombolítica) o por la instrumentación endovascular encaminada a la reperfusión de la ARI.145 De hecho en un análisis donde se comparó ACTP vs ACTP + STENT no se demostró mayor beneficio con esta última estrategia, inclusive existió tendencia a mayor mortalidad (3.1% con balón 5.8% con STENT, p < 0.07), lo que se adjudicó a menor frecuencia de obtenerse TIMI 3E con STENT.146,147 Hecho que a su vez puede sugerir mayor grado de microembolización miocárdica. Los principales megaestudios más recientes como el CADILLAC, el ASSENT-3, el GUSTO - V y el ADMIRAL han demostrado con STENT o sin este recurso endocoronario y la administración de IRPG, flujos TIMI-3E similares sin mayor impacto en la reducción en la mortalidad del IAM con ellos, pero sí beneficio de menor trombosis aguda y subaguda del stent o de la ARI.147,148,150,151 Tal observación ha resultado hasta cierto punto decepcionante, ya que se esperaba mejor o mayor impacto benéfico sobre la microcirculación coronaria al realizarse el bloqueo plaquetario con IRP IIb/IIIa. Esto se fundamentaba en hechos tales como el haber documentado mejoría en la contracción regional e incremento en la FE con el uso de ellos.152 Tampoco en el escenario del enfermo diabético con IAM han resultado favorables, a diferencia de lo que le ocurre al diabético con SICA SEST cuando se utilizan IRP IIb/IIIa, donde el beneficio clínico es palpable.143 Se ha visto en tratándose de diabéticos con IAM o efecto nulo o mayor frecuencia de sangrados importantes (ASSENT - 3, GUSTO - V).148,150 Lamentablemente como estrategia sólida establecida de agentes antiplaquetarios sólo está la utilización de ASA que sí ha demostrado que sola o en conjunto con la terapia fibrinolítica es capaz de reducir la mortalidad en el IAM.10,11 Mas no quiere decir esto que se esté negando el efecto noble que han demostrado los IRP IIb/IIIa sobre la arteria epicárdica, pero tampoco hay evidencia sólida de que tengan mayor impacto adecuado sobre la trombosis microcirculatoria en el escenario del IAM. Hay nuevas estrategias terapéuticas que están encaminadas a la utilización de dosis menores de trombolíticos con períodos más sostenidos de IRP IIb/IIIa y que van más orientadas a tratar de obtener mejor reperfusión microvascular. Por otro lado, no podemos ignorar que no sólo es la trombosis plaquetaria la responsable de los cambios en la permeabilidad microvascular, también está incluido el papel de la cascada de neutrófilos y los efectos posibles de la propia reperfusión microvascular y de la inflamación (Figs. 2 y 3).153
La respuesta inflamatoria en el IAM
El IAM está asociado siempre a una reacción inflamatoria, la que es prerrequisito para el proceso de curación y formación de la lesión final cicatricial.154 La oclusión coronaria crítica reduce el flujo coronario al área miocárdica involucrada modificando de manera significativa el metabolismo energético tisular. Sabemos que oclusiones breves de cinco minutos pueden dar como resultado anormalidades en el miocardio reperfundido que duran hasta 24 - 48 horas para recuperarse, por lo tanto que no son letales y que finalmente el tejido se recupera. Lo que ya ha sido analizado como miocardio aturdido. Estas alteraciones funcionales se han relacionado a la producción de radicales libres pero no claramente a procesos de índole o respuestas de carácter inflamatorio. En cambio si la isquemia se prolonga el proceso inflamatorio estará siempre presente, mismo que se ve reforzado si el tejido isquémico es reperfundido. La primera evidencia de que la inflamación agrava el daño miocárdico proviene del resultado de haberla observado después de aplicar estrategias antiinflamatorias en modelos animales de isquemia miocárdica y de reperfusión. La administración parenteral de corticosteroides demostró una clara reducción del tamaño del infarto en modelos caninos.155 Esta observación inicial dio origen al estudio de administración de metilprednisolona en enfermos con IAM lo que resultó en una verdadera catástrofe clínica: aumento de arritmias ventriculares malignas y de la extensión del área del infarto.156 Investigaciones posteriores demostraron categóricamente que los corticoesteroides disminuían el proceso inflamatorio al reducir el número de leucocitos infiltrados, pero retrasaban considerablemente la curación, la cicatrización y el depósito de la colágena. Esta observación ha recibido apoyo al demostrarse que la reperfusión mejora la reparación tisular (en su cara favorable) y que este efecto está mediado a su vez al desencadenarse la propia respuesta inflamatoria. Por lo tanto, es menester conocer mejor hoy día los eventos celulares y los moleculares que están asociados al binomio isquemia - reperfusión con miras a buscar la mejor estrategia terapéutica balanceada. En la que mitigue la inflamación y su daño durante la reperfusión temprana pero que no interfieran con el proceso de curación y el de cicatrización normal (Fig. 2). Se han realizado estudios experimentales que han tendido a guardar estos miramientos específicos de disminuir la respuesta inflamatoria en el IAM: reduciendo la generación de factores quimiotácticos al depletar el complemento, administrando inhibidores de la lipo-oxigenasa y antagonistas del leucotrieno B4, con los que se ha logrado reducir la zona del infarto. También se ha alcanzado disminuir la sensibilidad a la isquemia y abatir la zona infartada en modelos animales al emplear anticuerpos antineutrófilos, antimetabolitos que los depletan, o por medio de filtros que reducen las cantidades de los glóbulos blancos. Efectos similares se han obtenido al utilizarse antirradicales libres. Los aspectos profundos de lo complejo de esta temática de la inflamación en el IAM escapan de esta revisión pero pueden ser captados por el lector en una excelente y muy reciente publicación hecha por Frangogiannis N G y colaboradores.153 Es sólo nuestro interés hacer énfasis que éste es uno de los aspectos muy importantes modernos, conocidos parcialmente, de los que puede depender el obtener flujo miocárdico óptimo después de alcanzarse el flujo TIMI 3E. Brevemente señalaremos que el proceso de la inflamación está desencadenado por la activación del complemento. Así fue demostrado por Hill y Ward en un modelo experimental de infarto en ratas en el año de 1971.158 Posteriormente Pinckard y colaboradores159 consignaron que al ocurrir la necrosis miocárdica, daba origen a la liberación de constituyentes de la membrana subcelular ricas en mitocondrias, las que son capaces de activar los componentes de la cascada del complemento (C1, C2, C3 y C4). Otras observaciones posteriores han hecho plausible la idea de bloquear la activación del complemento, consumiéndolos o bien interfiriendo contra ellos por medio de anticuerpos. El papel de los radicales libres, de la cascada de las citoquinas, de los neutrófilos, de éstos con las selectinas y con el CD18 y beta dos integrelinas se están puntualizando por diferentes grupos de investigadores. Lo mismo los mecanismos por los cuales los neutrófilos producen o favorecen el daño miocárdico, al igual el papel modulador de la interleuquina en relación a la respuesta inflamatoria y el de los mastocitos en la formación de la cicatriz miocárdica, el de los fibroblastos y el proceso de la remodelación de la matriz extracelular.153 Pero la pregunta clínica importante ¿cuál es el estado actual de las estrategias terapéuticas antiinflamatorias en el IAM? Aunque la importancia de la cascada inflamatoria ha sido reconocida y estudiada en los últimos 25 años y hay una lista vasta de participantes en ella en el IAM, en los estudios experimentales en animales se demuestra una reducción del tamaño del IAM utilizando estrategias antiinflamatorias específicas, en el hombre infartado en general no han sido exitosas. Por lo tanto, es una causa aparentemente tardía de no alcanzar perfusión miocárdica óptima y para la cual no tenemos armas terapéuticas para revertirla en la clínica contemporánea en el IAM.
De lo experimental-a la epidemiológica clínica: anormalidades inmunológicas en el IAM transmural
Además de la numerosa experiencia experimental en este terreno y que ya fue consignada previamente en esta revisión en relación a los neutrófilos y su función anormal, en el IAM transmural existen también hechos de orden epidemiológico que dan soporte al papel que tiene el sistema inmunológico en enfermos con isquemia y con daño post reperfusión. Así lo asientan las observaciones hechas en las cuentas totales de glóbulos blancos (CTGB) y las realizadas en el análisis de la Proteína C reactiva en el IAM. Los estudios epidemiológicos han demostrado que la cuenta total de glóbulos blancos (principalmente integrada por los neutrófilos) ha resultado un factor pronóstico independiente de riesgo para la cardiopatía isquémica.160 Ésta correlaciona con el grado de enfermedad coronaria y el riesgo de reinfartos en los sobrevivientes del IAM. En estos sujetos aumenta la CTGB después del inicio del dolor, para alcanzar su pico máximo de 2 - 4 días más tarde y retornar a lo normal aproximadamente a la semana.161 En aquellos tratados con estreptoquinasa se han visto reducciones de la CTGB.162 En relación con la proteína C reactiva en sobrevivientes del IAM, ésta correlaciona con el desenlace de mortalidad a seis meses. Una disminución del pico de la proteína C reactiva, en este escenario del IAM se ha documentado cuando se obtiene permeabilidad óptima de la ARI, sugiriendo que la reperfusión efectiva reduce la inflamación en el IAM. También la proteína C reactiva se ha informado colocalizada con fragmentos activados del complemento en el seno del IAM en individuos que fallecieron por esta causa.163 Se le considera no sólo un marcador de la actividad circulante proinflamatoria de las citoquinas, sino también se sabe que puede contribuir a la inflamación en el miocardio activando el complemento.164 Se ha postulado que la aspirina reduce la síntesis de la proteína C reactiva, que impide su ligadura con las membranas y que posiblemente inhibe la activación de la clásica vía del complemento. Otro medicamento que abate la proteína C reactiva son las estatinas y el riesgo es tres veces mayor con enfermos con elevación de ésta al compararlo con placebo. Hay hallazgos que sugieren que la pravastatina puede atenuar los efectos inflamatorios que representan riesgo mayor coronario post IAM.165
SICA sin elevación del segmento ST
En este grupo de enfermos o al menos en la mayoría de ellos el bloqueo de receptores plaquetarios y por lo tanto el tratamiento de la microembolización parece mucho más alentador en su impacto terapéutico gracias a los conocimientos adquiridos en relación a los biomarcadores de necrosis, concretamente las troponinas ya que al contar con éstas le permiten al clínico decir cuales son los enfermos donde ha ocurrido la microembolización.3,4,6,8,119,166 Aquéllos con Troponina T con valores de 0.01 a 0.63 ng/mL son los que tienen mayor mortalidad y posibilidad de reinfarto (5% y 12% respectivamente) y éstos son en los que se puede inferir que tienen a su vez mayor propensión a la microembolización. Están en este grupo los que más se benefician del uso de IRP IIb/IIIa.5,6 Muy interesante resulta mencionar que los enfermos agrupados a su ingreso como anginas inestables o infartos no Q y que tienen cifras de Troponina T > 0.63 ng/ mL, que por lo tanto se acercan más al terreno del infarto Q, el impacto de los IRP IIb/IIIa no parece tan claro en ser favorable. Es muy novedosa la observación que en el otro extremo de los valores, que para algunos se consideran normales de Troponina T < 0.4 los IRP IIb/IIIa tienen muy probablemente beneficio clínico. El excluir esta población de su utilización marginaría un 10 -15 por ciento de los enfermos con SICA SESST de su utilidad.5 Es posible sin poder asegurarlo que tal vez el impacto de esta medicación sea más sobre la arteria epicárdica manipulada y menos sobre la microvasculatura comprometida, pero es plausible también que puedan prevenir o limitar los efectos de la microembolización producto de los PCI.119,138,152 Un aspecto a considerar es que aquellos enfermos que tienen embolización a la microcirculación deben tener una placa aterosclerosa muy activa con un componente inflamatorio relevante, lo que puede sugerir el hecho de haberse documentado interdependencia entre las Troponinas y elevaciones simultáneas de proteína C Reactiva.140 Por ende los enfoques terapéuticos futuros deberán encaminarse y agregar medicación anti-inflamatoria en estos sujetos y validarse en ensayos clínicos.
La valoración de la perfusión microvascular
Varios métodos se han utilizado para la valoración cuantitativa de la perfusión microvascular, lo que nos da una información muy valiosa de lo que está aconteciendo en el tejido miocárdico postpermeabilización de la ARI (Figs. 11 A y B). Estos procedimientos diagnósticos serán mencionados a continuación.
La resolución temprana de la elevación del segmento ST
En los enfermos con infarto agudo del miocardio o en evolución, contar con un indicador pro nóstico temprano, que nos oriente hacia la resolución favorable del proceso patológico sería altamente deseable en la clínica, particularmente después de haber empleado un método de reperfusión (terapia trombolítica o PCI). Aunque el ECG de superficie es nuestro primer instrumento diagnóstico y nos permite definir en qué magnitud se ha elevado el segmento ST, el observar la resolución temprana de la misma nos hablaría en cierta manera de una evolución favorable. Este hecho ha sido analizado en pocos estudios como un indicador de mortalidad o del pronóstico a corto plazo.167-168 En cambio el impacto que tiene la reperfusión temprana valorada por modificaciones del segmento ST ha sido investigada por varios grupos de investigadores.167-70 En la mayoría de los estudios una reducción del segmento ST de > 70% y menor de 30% son los puntos de corte electrocardiográficos más efectivos para poder predecir el tamaño del infarto, la función ventricular izquierda, así como la sobrevida a corto y largo plazo. En el estudio INJECT (The International Joint Efficacy Comparison of Thrombolytics) se realizó un subanálisis de 1,900 enfermos con IAM que habían recibido tratamiento trombolítico. Se consideraron tres tipos de resoluciones del segmento ST. Completa > 70 %, parciales de 30 a 70% y sin resolución < 30 a cero%. En 1,398 enfermos que habían acudido con menos de seis horas de evolución del infarto la resolución de la elevación del segmento ST fue completa, parcial y nula y la mortalidad documentada en cada condición fue de 2.5%, 4.3 % y 17.5 % (p< 0.0001).171 Cuando las características basales fueron tomadas en consideración, la resolución del segmento ST fue el principal factor independiente del pronóstico de mortalidad a 35 días. Por lo tanto la ausencia de resolución del segmento ST indica falla en la reperfusión y predice una mortalidad elevada (ausencia de TIMI 4 miocárdico). En cambio si ésta es completa se asociará a infartos de tamaño pequeño, a baja mortalidad y presumiblemente a mayor probabilidad de tener perfusión epicárdica y microvascular. Por lo tanto el ECG de superficie, tan a la mano en la clínica, analizado como lo sugiere Schroeder y sus colaboradores171 puede ser un instrumento valioso que indirectamente nos permite valorar el impacto de la reperfusión postintervención lítica y mecánica endovascular coronaria y especular sobre el pronóstico a corto plazo en el IAM.
La angiografía coronaria
Aunque la valoración de la calidad del flujo coronario epicárdico por este método es semicuantitativo, no hay duda que en la mayoría de las ocasiones es la angiografía coronaria nuestro primer instrumento diagnóstico certero que nos informa acerca del estado de permeabilidad de la arteria epicárdica postreperfusión. El grado de perfusión de la arteria epicárdica lo establecemos a través de la escala de flujo TIMI, el cual es un sistema útil, mas es subjetivo y con ciertas limitantes inherentes al propio método que ya han sido comentadas con antelación. Para conocerlo mejor, y darle mayor solidez a la información angiográfica, se ha creado un índice al respecto que analiza el número de cuadros del cine que requiere el material de contraste para alcanzar la circulación coronaria distal (The TIMI frame count).126,128,129 Este índice angiográfico se considera objetivo, cuantitativo, reproducible y sensible a los cambios del flujo coronario. En un modelo multivariado, donde se examinaron 1,248 enfermos que fueron analizados con este método, a los 90 minutos se encontró que como un factor pronóstico independiente capaz de predecir la mortalidad intrahospitalaria (RR: 1.21 por aumento de flujo en las 10 primeras imágenes, p < 0.001). Esta relación linear entre el flujo coronario a los 90 minutos y la mortalidad, también se documentó a los 30 días para los puntos compuestos cardiovasculares de: muerte-reinfarto-ICC y fracción de expulsión < 40%.130 La utilidad radica en que también permite identificar a los sujetos con un bajo riesgo cardiovascular, así, ninguno de los enfermos del estudio GUSTO con una cuenta < 14 a los 90 minutos y sometidos a tratamiento fibrinolítico falleció.129 Tampoco se documentó mortalidad alguna en el estudio RESTORE (Randomized Efficacy Study of Tirofiban for Outcomes and Reestenosis) en los sometidos a ACTP que tuvieron esta respuesta del flujo coronario.32
Constituye el grado de flujo coronario que se le ha designado TIMI 3M y que traduce una respuesta que es superior al 95% de flujo coronario normal (Fig. 14). Más recientemente se ha creado otro método angiográfico simple. Los autores lo designan como "sombreado miocárdico" o "TIMI myocardial perfusion blush grade", que evalúa tanto la llegada del material de contraste como su vaciamiento en el miocardio.126,128-130 La valoración de la perfusión miocárdica por este método se cataloga de acuerdo a la siguiente es cala: grado TIMI 0M: sin perfusión miocárdica, TIMI 1M: presencia del sombreado miocárdico pero no eliminación del mismo de la microcirculación, TIMI 2M: sombreado miocárdico pero desaparición muy lenta, TIMI 3M: sombreado en la microvasculatura con lavado y desaparición del contraste en tres ciclos cardíacos. Este método ha permitido una estratificación adicional a la que proporciona el análisis del flujo TIMI epicárdico en relación a la mortalidad. En el estudio TIMI 10 B se obtuvo la siguiente información: con TIMI 3E y TIMI M 0-1: se observó letalidad del 10.9%, con TIMI 3E y M2: del 4.4 % y con TIMI 3E y TIMI 3M la mortalidad fue de tan sólo: 0.7%. 130 Esto claramente traduce que lo relevante de alcanzar "mas allá " del TIMI 3E de perfusión es la óptima irrigación microvascular, o sea el flujo TIMI 4 MIOCARDICO (TIMI 3E +TIMI 3M). Aunque el análisis del flujo angiográfico coronario nos da información valiosa, su profundidad diagnóstica es limitada en relación a la especificidad del método para conocer el verdadero estado de la microcirculación coronaria postreperfusión de la ARI. Al respecto analizaremos otras técnicas o métodos que nos dan más detalles, algunas de naturaleza obligadamente invasiva y otras no intervencionistas, que son las que lucen con mayor aplicabilidad futura en la clínica de esta patología.

Los estudios del flujo con Doppler intracoronario
Clásicamente, el flujo coronario es profuso durante la diástole ventricular con caída del mismo durante la sístole cardiaca, donde la microcirculación determina la magnitud del flujo a este nivel por el mecanismo de autorregulación. De existir obstrucción microvascular por embolización (microtrombo plaquetario, detritus de la matriz de la placa) o injuria microvascular por espasmo o daño postreperfusión, la suma del área de la sección transversal del lecho microvascular coronario se encuentra disminuida, lo que a su vez ocasiona reducción de la velocidad del flujo coronario epicárdico. En enfermos infartados con daño microvascular, se ha documentado una disminución de la relación diastólica/sistólica de las velocidades del flujo coronario y también de la reserva coronaria. Así, en sujetos con TIMI E2 y 3 se ha observado: flujo anterógrado sistólico disminuido, flujo anormal sistólico temprano retrógrado y desaceleraciones rápidas de la velocidad del flujo diastólico.172,173
A diferencia de lo que se documenta en el miocardio aturdido, en el daño tisular miocárdico irreversible los estudios con Doppler intracoronario muestran un patrón que se caracteriza por franca disminución del flujo basal, que no mejora durante la fase de hiperemia con el tratamiento farmacológico.174 Cuando se documenta postreperfusión, un patrón normal del Doppler intracoronario refleja razonablemente que hay un daño mínimo en la microcirculación o que está ausente, en esta condición la probabilidad de desarrollar insuficiencia cardíaca es de < 1%. En cambio si el patrón del flujo coronario obtenido por Doppler inicialmente es anormal y mejora al séptimo día, la expectativa que aparezca ICC es tan alta como del 6 al 21% de acuerdo a las observaciones de Brochet y coautores y Sakuma y colaboradores.175,176
Los estudios de medicina nuclear
Esta metodología moderna con imágenes de fotón único con talio o con tecnecio (nos da informes acerca de la integridad de la membrana celular o datos en relación de la función mitocondrial) son modalidades que tienen un verdadero valor acerca del estado de la perfusión microvascular coronaria (Fig. 13). Schoefer y colaboradores84 han sido de los primeros en demostrar anomalías en la perfusión tisular con estos métodos diagnósticos en individuos con IAM. Kondo y coautores177 en sujetos que habían alcanzado flujo coronario TIMI 3E examinaron la microvasculatura utilizando macroagregados de albúmina-tecnecio 99 m administrado de forma intracoronaria y demostró que en el 32% de ellos habían defectos perfusorios. Aquéllos con anomalías circulatorias distales demostradas con talio-201 tenían función ventricular izquierda regional deprimida. También con tomografía por emisión de positrones (PET) a los 30 días se corroboraron estos defectos en el miocardio. Con PET se han demostrado estas alteraciones del flujo en la microcirculación coronaria y en los aspectos concernientes a viabilidad tisular utilizando rubidio 82, NH3 y F-2 desoxiglucosa. Por lo tanto hoy día se sabe que es factible con imágenes de emisión de fotón único, obtenidas en tomografías o con PET, identificar sujetos con anomalías en la perfusión microvascular pero que tengan viabilidad tisular, mismos que se llegan a beneficiar con la terapéutica dirigida en especial a ese territorio del miocardio potencialmente recuperable.178,179
La resonancia magnética nuclear
Varios autores han investigado el potencial diagnóstico que tiene la resonancia magnética nuclear en caracterizar las imágenes de las zonas de infarto, las de la isquemia y por lo tanto las de la perfusión miocárdica microvascular alterada. En este terreno es clásico el estudio de Wu y colaboradores131 en sujetos con IAM y flujo TIMI 3E, en quienes identificó defectos perfusorios utilizando este método. Demostró que aquéllos con defectos miocárdicos en la resonancia tenían una probabilidad del 45% de tener eventos cardíacos mayores vs 9% si éstos estaban ausentes y a las 25 semanas de seguimiento se observó en los primeros menor sobrevida. Más aún, catalogó la magnitud de los defectos con la posible frecuencia de que acontecieran eventos cardiovasculares indeseables en estos enfermos. De ser pequeños, la sobrevida libre de eventos cardíacos fue del 82%, en los medianos del 70% y con imágenes anormales mayores en menos del 70%. Si bien esta técnica es sofisticada y particularmente onerosa, es posible que con el devenir del tiempo se torne más accesible para la clínica.180-182
La ecocardiografía contrastada miocárdica [ECM ]
Es una de las técnicas más prometedoras para darnos información acerca del estado de la microcirculación coronaria y para conocer su estado después de haber aplicado un método de reperfusión en la ARI en la práctica clínica hoy día. Los primeros estudios con esta modalidad diagnóstica se realizaron en las salas de cateterismo cardíaco, mediante la infusión intracoronaria de microburbujas de material sonicado, demostrándose defectos perfusorios miocárdicos y por lo tanto el área en riesgo después de la permeabilización de la ARI o bien consignándose la normofuncionalidad del territorio miocárdico.85,176,183,184 Lo relevante es que hoy día es posible obtener estas imágenes a la cabecera del enfermo en un tiempo relativamente corto (Fig. 15). Las observaciones iniciales en este sentido demostraron que existe una estrecha correlación entre el grado de TIMI E y los patrones de perfusión miocárdica obtenidos con la EMC.84,85 En un estudio realizado en 86 enfermos que recibieron dentro de la ventana útil de reperfusión y que alcanzaron TIMI 2E, todos tuvieron patrones de perfusión microcirculatorio anormales con la ECM. En cambio en aquéllos con TIMI 3E se documentaron defectos sólo en el 16%. Este grupo con imágenes anormales aun en presencia de TIMI 3E presentaron función ventricular abatida con poca recuperación a los 28 días y el reestablecimiento de la función ventricular en la clínica fue equiparable al del grupo con IAM y TIMI 2E.183,184 Otras investigaciones ulteriores han corroborado estos hechos documentados inicialmente. La ECM es un método que indica de una manera cuantitativa o global el estado de la perfusión microvascular y puede ser un indicador de la recuperación funcional y clínica del enfermo con IAM. De las diferentes técnicas ecocardiográficas modernas y de las más prometedoras es la ECM con la caracterización ultrasónica del tejido miocárdico en la que se utiliza material sonicado de contraste (microburbujas de perfluorocarbono) inyectadas por vía endovenosa periférica. La caracterización tisular del tejido miocárdico con esta técnica (integrated back-scatter (IBS)) ha sido empleada en el IAM y se ha demostrado que es dependiente del ciclo cardíaco y que la imagen de densitometría acústica está eliminada o atenuada en el tejido miocárdico isquémico o en el infartado al ser comparada con la imagen que da el tejido preservado.185,186 También se ha notado que la recuperación del tejido miocárdico reperfundido con éxito, estudiado en el séptimo día, se asocia a mejoría en la contracción regional, lo que sugiere viabilidad microvascular postreperfusión de la ARI. Takiuchi y coautores187 han confirmado esta observación tan relevante en enfermos con IAM tratados con PCI. Los investigadores consignaron que, en la zona afectada, al tercer día había mejoría de la caracterización ultrasónica del tejido cardíaco, mas sin asociarse con beneficio en los índices de contracción regional ventricular, pero sí notándose efecto favorable a los 21 días de observación. En un trabajo experimental en perros realizado por Leistad y colaboradores,188 en el que efectúan desde la obstrucción completa coronaria a diferentes grados de estenosis de la arteria descendente anterior, los autores demuestran modificaciones en la configuración de las curvas de perfusión de densitometría acústica. Observaron desde ausencia del flujo coronario hasta caídas significativas de la curva de densitometría en relación con el tiempo. El método ha demostrado una buena correlación con el flujo coronario medido con microesferas radioactivas en relación a la imagen de video densitometría (r:0.83, p < 0.0001). Mas esta observación experimental debe aún confirmarse en humanos con cardiopatía isquémica y lesiones coronarias significativas y en aquéllos con obstrucciones de poca cuantía. Estas impresiones sólo se han recabado en grupos reducidos de enfermos y hay que estar conscientes que hay ciertas limitaciones con esta técnica y que aunque los datos ecocardiográficos son fáciles de colectar en manos expertas su análisis es y luce más complicado. La orientación de las fibras miocárdicas profundas y lo distal del campo a explorar afectan significativamente la imagen de la caracterización ultrasónica. En el eje corto el septum interventricular anterior tiene los más altos niveles de caracterización ultrasónica, donde el haz es prácticamente perpendicular a las fibras miocárdicas. En cambio la pared lateral del ventrículo izquierdo y el septum posterior son las que tienen los niveles más bajos, donde la orientación de las fibras miocárdicas es paralela al haz del transductor del ultrasonido. La pared posterior resulta la de menor reflejo acústico por su profundidad. Por lo tanto la anisotropía cardíaca debe ser corregida en esta técnica. Aún con estas limitaciones dadas por la magnitud de la profundidad de las células cardíacas, la orientación de las fibras miocárdicas y las variaciones propias que se han observado en sujetos sanos, la ECM luce relevante. Una comunicación muy reciente de la capacidad de poder determinar el éxito o no de la reperfusión de la ARI, después del empleo del tratamiento fibrinolítico, utilizando la caracterización tisular ultrasónica del miocardio es la información que se desprende del trabajo de Hancock y colaboradores.189 En el estudio de 113 enfermos con IAM sometidos a trombólisis y donde se realizó validación angiográfica del flujo coronario se demostró que utilizando la diferencia en los picos de la caracterización ultrasónica (integrated back-sacatter (IBS) ) del miocardio anormal y sin ARI permeable vs el tejido infartado, mas reperfundido, que el IBS era mucho más bajo en la zona infartada y no irrigado o con arteria ocluida postfibrinólisis, que la del tejido que sí fue reperfundido con éxito postrombólisis (3 .3 vs 4.6 dB, p < 0.00001). Los autores determinaron un valor de corte del 15% (1.5 dB) entre el segmento infartado y el normal, siendo la sensibilidad, la especificidad, el valor predictivo positivo y negativo para conocer si se obtuvo permeabilidad de la ARI post-reperfusión de 92%, 75%, 81% y 89%, respectivamente. Esta información es trascendente, ya que el método no es invasivo a diferencia de la arteriografía coronaria, que además requiere de facilidades del laboratorio de cateterismo, mismas que no existen hoy día en todas las Instituciones de los países desarrollados o en vías de ello (Fig. 16).


¿Es modificable el fenómeno de no flujo consecuencia de la microembolización y del daño postreperfusión?
Con toda la evidencia experimental y la clínica mencionada previamente de la patofisiología de la cardiopatía isquémica aguda, queda poca duda de que en el tratamiento moderno de los síndromes coronarios agudos con o sin elevación del segmento ST, es muy relevante enfocar nuestra atención terapéutica a la microcirculación coronaria. Esto amén de la ya establecida y sustentada sobre la importancia de obtener permeabilidad óptima de la arteria epicárdica. La investigación sobre el tratamiento del fenómeno de microembolia plaquetaria vinculado a la obstrucción microvascular con terapia antiplaquetaria ha dado resultados sólo prometedores. En cambio para aquéllas encaminadas hacia la corrección de la injuria postreperfusión los resultados parecen mucho más alentadores en el terreno experimental, mas lamentablemente ésta no lo es en el escenario clínico. Los ensayos con inhibidores de los receptores de la glucoproteína plaquetaria IIb/IIIa realizados in vitro e in vivo, han apuntado que con su utilización es posible mejorar la disfunción microvascular producto de la microembolización plaquetaria. Se ha demostrado que con su empleo se reduce la masa del trombo epicárdico y la plataforma para la generación de trombina, se favorece la penetración del trombolítico, se disminuyen los inhibidores de la trombólisis (alfa 2 inhibidor plasmático y PAI -1) e inhiben los receptores plaquetarios IIb/IIIa con lo que se impide la agregación de estos elementos formes de la sangre al reducir además la viscosidad de las mismas. Con el uso de ellos se ha visto que los agregados plaquetarios constituidos son más débiles que cuando se administra placebo o sólo heparina, lo que limita la agregación y dan mayor exposición a la fibrina, por lo tanto favorecen la acción de los fibrinolíticos exógenos. De manera sucinta podemos decir que, además de favorecer la permeabilidad de la arteria epicárdica, estos agentes actuarían distalmente en la microcirculación evitando la microembolización y la trombosis (Figs. 17 A y B).190 Con esta medicación se ha consolidado el concepto que se favorecen con mayor frecuencia flujos TIMI 3E a los 90 minutos. Así ha quedado demostrado, al comparar estrategias de fibrinolíticos o de la ACPT sola, y cuando se ha empleado además el inhibidor del receptor plaquetario. En diferentes megaestudios utilizando abciximab, tirofiban o eptifibatide como son: el TIMI-14, el PAMI, el PAMISTENT, el CADILLAC, el GUSTO V, el ADMIRAL y el ASSENT 3 se han logrado obtener en promedio flujos coronarios TIMI 3E en el 90-95% en los enfermos a los 90 minutos.147,148,190 Con esta mejoría del flujo coronario epicárdico se esperaría un gran beneficio en reducir la mortalidad del IAM. Se ha demostrado con abciximab en el estudio ADMIRAL una reducción de puntos cardiovasculares adversos hasta del 80% a los 30 días comparado con el grupo control (p < 0.031).151 También se han colectado datos que permiten afirmar que existe menor trombosis de la ARI, del stent y se favorece la función ventricular. Si estas mejorías en el flujo epicárdico coronario es mediada directamente por la inhibición de la agregación plaquetaria o por el reestablecimiento más rápido de la permeabilidad de la ARI o por beneficio directo en la microcirculación coronaria aún está por establecerse en la clínica. Lo importante a consignar es que el impacto que se esperaba que darían al utilizarlos de manera asociada con la fibrinólisis o los PCI, permitía pronosticar una reducción cercana al 2% en mortalidad del IAM, condición que no se ha alcanzado en los estudios antes citados a pesar de haberse incrementado la tasa de reperfusión de la ARI en un 20- 25%.147,148,151,190 Deseamos recordar que hay otras observaciones clínicas, como aquellas que documentan flujos TIMI 3E + TIMI 3M, con mortalidad cercana al 1.5% para el IAM. En el estudio GUSTO II b sólo con ACTP sin bloqueador del receptor plaquetario, se alcanzó una mortalidad del 1.6% con TIMI 3E. En cambio en el estudio CADILLAC en enfermos con IMA, que en un 88% estaban en clase Killip-Kimball I, la mortalidad con ACTP + abciximab fue de 2.5% y con stent + abciximab del 4.2% a los seis meses.147, 148 En el estudio GUSTO V (fibrinólisis + abciximab), la mortalidad global fue del 6%, aunque es de mencionar que ha sido una de las letalidades más bajas documentadas en la historia del IAM con terapia trombolítica. Mortalidades similares se han documentado en el estudio ASSENT-3, donde se utilizaron como estrategias para el tratamiento del IAM: tenecteplase con enoxaparina (5.4%), asociada a abxicimab (6.6%) y con heparina no fraccionada (6.0%).150 Aunque recordaremos que cuando se utiliza esta modalidad de reperfusión y se alcanza TIMI 3E, la mortalidad que se ha anotado es del 4.4%. Otra observación importante es la que se deriva del metaanálisis de estos medicamentos en los SICA sin elevación del ST, donde éstos han logrado abatir 1% la mortalidad de manera absoluta.143 Además demuestran claro beneficio en los enfermos diabéticos, sean enviados o no a procedimientos de reperfusión mecánica. En cambio al realizar el análisis del grupo de diabéticos con IAM, los bloqueadores de los receptores plaquetarios o no tienen efecto o resultan deletéreos. Así ha quedado documentado en el GUSTO V y en el ASSENT 3.148,150 Lamentablemente se tiene que decir que aparentemente aunque hay claro beneficio con el empleo de bloqueadores de los receptores plaquetarios en la arteria epicárdica, no se ha logrado demostrar que lo mismo acontece con su utilización a nivel de la perfusión microvascular en el IAM (Figs. 17 A y B). Otros caminos se han trazado para resolver la problemática de la microembolización por medio de dispositivos mecánicos de aspiración o con el uso concomitante de filtros o cestillas, instrumentos hemodinámicos que hoy día están en plena evaluación clínica. El catéter de trombectomía X-sizer se ha diseñado para la remoción selectiva del trombo y de los detritus de la placa antes de colocarse las mallas endocoronarias (Fig. 18). El llamado sistema Guardwire que consta de un balón, mismo que se infla distalmente a la lesión oclusiva y que va encaminado a reducir la microembolización y que ha sido sólo ensayado en lesiones que aparecen en injertos venosos safenos. Sin embargo estos dispositivos no se han instrumentado aún en el IAM donde la porción distal a la obstrucción no se visualiza a priori.147 Toda esta información hay que tomarla con extrema cautela, ya que es posible que sólo estén actuando a solucionar uno de los posibles factores que participan en el deterioro de la misma, faltaría modificar los efectos de la injuria postreperfusión, posiblemente los relacionados con el rubro del espasmo microvascular y los de la inflamación en el IAM.


Por lo tanto hay otras estrategias en marcha tratando de resolver estas importantes interrogantes. En relación al componente microespástico se ha utilizado la papaverina, la adenosina, el verapamil y el nicorandil, este último un nitrovasodilatador recién ensayado en la angina de pecho en el Japón y en Europa.191-193 Éste parece ofrecer cierta protección contra los efectos deletéreos de la mancuerna isquemia-reperfusión. Los estudios han demostrado que con su infusión se reduce el tamaño del infarto, se mejora la perfusión microvascular y se aumenta la magnitud del miocardio salvado. El mecanismo por el que actúa no está claro, pero parece estar ligado a sus acciones de reducir la pre y postcarga ventricular, además de su efecto antineutrófilo y antirradical libre que tiene este nitrato que sensibiliza los canales de ATP-potasio.194 Se han hecho también esfuerzos por reducir la lesión postreperfusión inhibiendo la activación y el acúmulo de los neutrófilos. De los IECA, con el cilazaprilat se ha demostrado en modelos experimentales animales que atenúa la adhesión y activación postisquémica de los neutrófilos. Enfermos tratados con HMG-Co A reductasa como la lovastatina y la simvastatina se ha avalado que dan reducción de la expresión del CD11b de superficie y de la adhesión de los monocitos al endotelio.194 Datos que dan soporte a la hipótesis que parte del beneficio atribuible a las estatinas, además del conocido hipolipemiante, es debido a la inhibición de los neutrófilos.165 Experimentalmente ha sido valiosa la observación que el empleo de anti CD 18 b reduce el área de infarto y de no flujo en animales.195 Lo que ha dado origen al estudio clínico LIMIT-AMI de fase II, en el cual se estudiará la seguridad y eficacia de la administración de anti CD 18 b en sujetos con IAM sometidos a tratamiento con t-PA. La adenosina, en el terreno experimental, también ha puesto en evidencia que incrementa el flujo coronario y que reduce el tamaño del área infartada. Observación que dio origen al estudio AMISTAD I (Acute Myocardial Infarction Study of Adenosine) y donde se demostró en el IAM de la cara anterior, mas no en el de la pared inferior, una reducción del 70% del tamaño del infarto. Lo que se asoció a mayor número de flujos TIMI 3E y menor frecuencia de fenómenos de no flujo, con mejoría en la recuperación de la movilidad de la pared ventricular, función ventricular global y evolución clínica favorable.196 Actualmente se encuentra en marcha el estudio AMISTAD II (fase III) que se llevará a cabo en 2,100 enfermos con IAM de la cara anterior en donde se compararán dos dosis de adenosina intravenosa vs placebo más terapia adjunta de reperfusión.
Los agentes antitrombóticos son una parte muy importante de la terapia de reperfusión medicamentosa en el IAM. Hoy día la aspirina y la heparina no fraccionada son suministradas de manera rutinaria a la mayoría de los enfermos con esta variedad de SICA. Sólo reciente es la utilización de la heparina de bajo peso molecular y en particular su administración conjunta con la terapia fibrinolítica. A diferencia de la heparina no fraccionada la de bajo peso molecular tiene una cinética mucho más predecible, da menos trombocitopenia, se une menos a las proteínas y no requiere de monitorización de la coagulación. La mayoría de los estudios con heparinas de bajo peso molecular han demostrado con su utilización menor frecuencia de reoclusiones de la arteria responsable del infarto, mayor permeabilidad tardía, de reinfartos e inclusive han proporcionado datos que apuntan hacia una mejor perfusión tisular, al ser comparado con la heparina no fraccionada.150,190,197
Recientes son los resultados del estudio ENTIRE-TIMI 23 en el IAM donde se analizan y comparan los efectos de dos estrategias de reperfusión: tenecteplase (TNK) a dosis completa + heparina no fraccionada (HNF) vs enoxaparina (ENOXA) y la terapia combinada de 1/2 dosis de TNK + abciximab + HNF vs ENOXA. Los autores concluyen que tanto la HNF como la ENOXA dan igual frecuencia de flujos TIMI 3 E (50 y 51% respectivamente), sin embargo la frecuencia de puntos compuestos cardiovasculares de muerte-IAM recurrente fue de 15.9% para HNF y de 4.4% para ENOXA con menor frecuencia de episodios hemorrágicos para la heparina de bajo peso molecular. Los autores especulan que es posible que los efectos superiores de la ENOXA radican en dar menor frecuencia de trombosis epicárdica y que se pueda traducir en menor daño en la microcirculación coronaria. Sin embargo, aunque los resultados del estudio AS-SENT-3 y los del ENTIRE-TIMI 23 sugieren que la heparina no fraccionada puede ser substituida por la ENOXA, aún se requieren más estudios clínicos de fase III para contestar esta importante interrogante clínica.198
Otra terapéutica que está actualmente en investigación como tratamiento adjunto en el IAM es la utilización del pentasacárido (Estudio PENTALYSE) inhibidor selectivo del factor Xa. Los resultados iniciales sugieren que su asociación con fibrinolíticos (alteplase) es segura y efectiva como son las heparinas de bajo peso molecular y que representa un concepto atractivo para el tratamiento futuro del IAM.199
Los procedimientos coronarios intervencionistas "facilitados"
El Registro Nacional de los Estados Unidos de Norteamérica ha claramente demostrado que mientras más retrasado sea el tiempo puerta-balón se asocia a una mayor mortalidad del IAM. Los enfermos que son llevados con un retraso de 121 a 150 minutos tienen un aumento relativo de la mortalidad del 41% y de ser de 150 a 180 minutos el incremento relativo de la letalidad es de 62%. También se conoce que el tiempo puerta-fibrinólisis hoy día se ha disminuido de 61.8 a 37.8 minutos (p < 0.0001).200
Por lo que una limitante muy importante o mayor de la reperfusión mecánica hoy día es la falta de una logística fluida para concretarla, en cambio con la fibrinólisis tiende a acortarse. Esto ha dado origen a buscar una estrategia terapéutica mixta en el IAM, aquella que combine los conocimientos de la reperfusión medicamentosa con la intervencionista invasiva, lo que ha dado origen al concepto moderno de la reperfusión mecánica facilitada.147 La reperfusión medicamentosa está encaminada a dar lo más pronto posible el mejor flujo TIMI E con miras a realizar a seguir los PCI y alcanzar flujo TIMI 3E si éste no se ha obtenido (Figs. 19 A, B, C y D). También está particularmente encaminada a resolver la inestabilidad de la placa ateroesclerosa rota o fisurada. Esta estrategia a su vez permite remover el excedente trombótico y ateroscleroso de la ARI (Figs. 18 y 19 B). Si en el pasado se había visto una evolución desfavorable en enfermos con SICA CESS con la utilización de los PCI a seguir de la fibrinólisis (angioplastía de rescate), hoy día sabemos que se pueden efectuar de una manera segura y efectiva.201-205 El estudio SPEED demostró que tanto la realización de los PCI postrombólisis o dosis reducidas de fibrinolíticos en combinación con bloqueadores de los receptores plaquetarios IIb/IIIa se asociaba a mejores resultados que con la sola utilización de la reperfusión farmacológica.205,206
Una observación muy importante realizada por los investigadores del PAMI y del Registro del Hospital Moses Cone es que aquellos sujetos con IAM enviados a ACTP primaria, que llegaban al laboratorio de hemodinámica con la ARI permeable vs ARI cerrada tenían mayor éxito del procedimiento intervencionista, menor tamaño del infarto, mejor recuperación de la función ventricular y menor mortalidad temprana y tardía.145,207 Estos importantes estudios establecen la posibilidad y la esperanza de que la estrategia combinada pueda dar mejores resultados que la reperfusión sola con trombólisis o únicamente con PCI, tanto en la arteria epicárdica responsable del IAM como a nivel de la microcirculación coronaria, como ha sido observado aisladamente en nuestro medio por ecocardiografía contrastada en el infarto del ventrículo derecho con la estimulación de dobutamina.208 Hipótesis que será validada en el estudio FINESSE que está actualmente en marcha. Esta sinergia terapéutica que se busca en la reperfusión del IAM es novedosa y esperamos que mejore los resultados obtenidos hasta la fecha y en particular que logre abatir el techo de mortalidad que se tiene hoy día en el IAM al obtenerse flujo coronario TIMI 3E y en especial TIMI 3M (Figs. 19 C y D).
Conclusiones
A pesar de las mejores opciones terapéuticas al ser aplicada la terapia de reperfusión en el escenario del IAM dentro de la ventana aceptada de seis horas, para la administración de fibrinolíticos o la aplicación de PCI (ACTP con o sin stents), un número no despreciable de enfermos quedan con un flujo coronario epicárdico inadecuado. Más aún el daño remanente en la perfusión microvascular luce más relevante, lo que a su vez indica un mal pronóstico y un elevado número de eventos cardíacos adversos por venir. Hay que aceptar que la terapia moderna para el tratamiento de este daño en la perfusión microvascular o disfunción de ésta, así como principalmente el que ocasiona la injuria postreperfusión no está resuelta hoy día.
Por lo tanto, el mirar o investigar qué existe "más allá de la reperfusión de la arteria epicárdica y de obtener flujo TIMI 3E" es relevante. Menester obligado es determinar si existe o no disfunción microvascular y conocer si existe daño tisular postreperfusión, lo que indicará una población que después de haber sufrido un IAM tiene un elevado riesgo de morbimortalidad cardiovascular. Al tener presente estos aspectos de la patofisiología moderna del IAM, cae por su propio peso la importancia de identificarlos de manera muy temprana en la clínica. Desafortunadamente no podemos decir que tenemos todas las armas de diagnóstico a la mano, pero sí que se encuentran en un verdadero auge tecnológico de desarrollo en esta época moderna de la cardiología. Particularmente cuando la información sobre la permeabilidad o no de la ARI la requerimos en los primeros 90 minutos para conocer si la reperfusión coronaria epicárdica se alcanzó con éxito. Información que obtenemos cuando los enfermos son enviados a procedimientos de reperfusión mecánica de la arteria epicárdica; sin embargo, bien sabemos que sólo en el 10% de los países desarrollados existen departamentos de hemodinámica o tienen estas facilidades las 24 horas. Por lo tanto las técnicas tales como la propia angiografía coronaria, el Doppler intracoronario, los estudios de medicina nuclear y la resonancia magnética nuclear no están fácilmente accesibles en la clínica para obtener esta información tan valiosa en relación de la permeabilidad de la arteria epicárdica y si existe o no disfunción microvascular o daño por la reperfusión. El estudio que más luce acercarse a darnos los elementos requeridos en esta ventana diagnóstica es la ecocardiografía miocárdica contrastada. Pero aún no existe una amplia experiencia al respecto y la tecnología a primera instancia no parece accesible a todos los medios hospitalarios. Sin embargo, la utilización de estas técnicas encaminadas a valorar la perfusión microvascular han sido la guía para buscar nuevos caminos terapéuticos para mejorar el estado del tejido miocárdico postreperfusión en el IAM. Los pasos clínicos en el tratamiento inicial del IAM quedan claros: obtener el mejor flujo coronario epicárdico (TIMI 3E), disminuir el daño microvascular y la injuria postreperfusión (TIMI 3M). Con el empleo de los bloqueadores de los receptores plaquetarios IIb/IIIa, se ha notado beneficio sobre la manipulación fibrinolítica o mecánica de la ARI y cierta atenuación de la obstrucción trombótica plaquetaria microvascular, lo que ha devengado beneficios en la función ventricular y al parecer mejores estados de evolución clínica post IAM. Mas no se ha demostrado de manera categórica una reducción en la mortalidad con ellos en los SICA con elevación del segmento ST, a diferencia del grupo sin supradesnivel de éste donde se ha abatido en forma absoluta uno por ciento la letalidad. Los esquemas terapéuticos ahora en investigación incluyen la combinación de fibrinolíticos a dosis bajas o a los PCI asociados a la terapia antiplaquetaria. También se investigan la utilización de la terapia directa antagónica de la trombina, las heparinas de bajo peso molecular y los pentasacáridos sintéticos, lo que puede dar un efecto aditivo en frenar la cascada de la coagulación. Finalmente la inhibición de los neutrófilos activados con anti CD18 y otros agentes parecen prometedores como armas terapéuticas para abatir el papel nefasto de estas células en el daño tisular postreperfusión. Hay sólidas premisas para enfatizar que en el tratamiento moderno del IAM debemos ver siempre que hay "más allá del flujo TIMI 3E" es decir: el flujo TIMI 4 ó MIOCÁRDICO.
Referencias
1. BRAUNWALD E, ANTMAN EM, BEASLEY JW, CALIFF RM, CHEITLIN MD, HOCHMAN JS ET AL: ACC/AHA guidelines for the managements of patients with unstable angina and non-ST segment elevation MI: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Patients With Unstable Angina ). J Am Coll Cardiol 2000; 36: 970-1062. [ Links ]
2. ANTMAN EM, BASSAND JP, KLEIN W, OHMAN M, LÓPEZ SENDÓN JL, RYDÉN L ET AL: Myocardial Infarction redefined-a cosensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959-69. [ Links ]
3. MORROW DA, CANNON CP, RIFAI N, FREY MJ, VICARI R, LAKKIS N: Ability of minor elevations of troponins I and T to predict Benefit From an Early Invasive Strategy in Patients With Unstable Angina and Non-ST Elevation Myocardial Infarction. Results From a Randomized Trial. JAMA 2001; 286: 2405-2412. [ Links ]
4. QUIN JM, MOLITERNO DJ: Troponins in Acute Coronary Syndromes. More TACTICS for an Early Invasive Strategy. JAMA 2001; 286: 2461-62. [ Links ]
5. LINDAHL B, DIDERHOLM E, LAGERQVIST B, VENGE P, WALLENTIN L, E INVESTIGADORES DEL FRISC II: Mechanisms Behind the prognostic Value of Troponin T in Unstable Coronary Artery Disease: A FRISC II Substudy. J Am Coll Cardiol 2001; 38: 979-86. [ Links ]
6. ANTMAN EM: TROPONIN MEASUREMENTS IN ISCHEMIC HEART DISEASE: More Than Just a Black and White Picture. J AM COLL CARdiol 2001; 38: 987-90. [ Links ]
7. ANTMAN EM, COHEN M, BERNINK PJLM, MCCABE CH, HORACEK T, PAPUCHIS G ET AL: The TIMI risk score for unstable angina-non ST-elevation MI: a method for prognostication and therapeutic decision-making. JAMA 2000; 284: 835-42. [ Links ]
8. MORROW DA, ANTMAN EM, SNAPINN SM, MCCABE CH, THEROUX P, BRAUNWALD E: An integrated clinical approach to predicting the benefit of tirofiban in non-ST elevation acute coronary syndromes. Application of the TIMI Risk Score for UA/NSTMI in PRISM-PLUS. Eur Heart J 2002; 23: 223-229. [ Links ]
9. LANGER A, GOODMAN SG, TOPOL E, CHARLES-WORTH A, SKENE AM, WILCOX RG, ARMSTRONG PW: Late assessment of Thrombolytic Efficacy (LATE ) Study: Prognosis in Patients With Non-Q wave Myocardial Infarction. J Am Coll Cardiol 1996; 27: 1327-32. [ Links ]
10. FIBRINOLYTIC THERAPY TRIALIST (FTT) COLLABORATIVE GROUP: Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomized trials of more than 1,000 patients. Lancet 1994; 343: 311-22. [ Links ]
11. ISIS COLLABORATIVE GROUP: Randomized trial of intravenous streptokinase, oral aspirin, both, or neither among 1,7187 cases of suspected myocardial infarction. Lancet 1988; 2: 349-360. [ Links ]
12. FARMER JA: Aggressive Lipid Therapy in the Statin Era. Progr Cardiovasc Dis 1998; 41: 71-94. [ Links ]
13. TIMI RESEARCH GROUP: Immediate vs delayed catheterization and angioplasty following thrombolytic therapy for acute myocardial infarction: TIMI II A results. JAMA 1988; 260: 2849-2858. [ Links ]
14. LENDERINK T, SIMOONS ML, VAN ES GA, VAN DE WERF F, VERSTRATE M, ARNOLD AER: Benefit of Thrombolytic Therapy Is Sustained Throughout Five Years and Is Related to TIMI Perfusion Grade 3 But not Grade 2 Flow at Discharge. Circulation 1995; 92: 1110-1116. [ Links ]
15. WEBER KT, CLARK WA, JANICKI JS, SHROFF SG: Physiologic versus pathologic hypertrophy and the pressure-overload myocardium. J Cardiovasc Pharmacol 1987; 10 ( Suppl. 6 ): s37-s50. [ Links ]
16. HOFFMAN JI: Heterogeneity of myocardial blood flow. Basic Res Cardiol 1995; 90: 103-111. [ Links ]
17. VETTERLEIN F, PRANGE M, LUBRICH D, PEDINA J, NECKEL M, SCHMIDT G: Capillary perfusion pattern and microvascular geometry in heterogeneous hypoxic areas of hypoperfused rat myocardium. Am J Physiol 1995; 268: H 2183-H 2194. [ Links ]
18. SCHEMERMUND A, BELL MR, LERMAN LO,RITMAN EL, RUMBERGER JA: Quantitative evaluation of regional myocardial perfusion using fast X-ray computed tomography. Herz 1997; 22: 29-39. [ Links ]
19. SCHELBERT HR: Cardiac PET; microcirculation and substrate transport in normal and disease human myocardium. Ann Nuclear Med 1994; 8: 91-100. [ Links ]
20. GAVIN BJ, MAXWELL L, EDGAR S: Microvascular Involvement in Cardiac Pathology. J Mol Cell Cardiol 1998; 30: 2531-2540. [ Links ]
21. REIMER KA, LOWE JE, RASMUSSEN MM, JENNINGS RB: The wavefront phenomenon of ischemic cell death I. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977; 56: 786-794. [ Links ]
22. SCHAPER W: Angiogenesis in the adult heart. Basic Res Cardiol 1991; 86 (Suppl, 2): 51-56. [ Links ]
23. HOFFMAN JI, SPAAN JA: Pressure-flow relations in coronary circulation. Physiol Rev 1990; 70: 331-390. [ Links ]
24. JANTUNEN E, COLLAN Y: Transmural differenes in ischaemic heart disease: a quantitative histologic study. Appl Pathol 1989; 7: 179-187. [ Links ]
25. HOYT JC, HUTCHINS GM: Variceal transformation of the subendocardial microvasculature in regions of chronic myocardial ischemia. Am Heart J 1989; 117: 830-836. [ Links ]
26. ZIELINSKI KW, KULIG A: Histomorphometric investigations of the human heart microvasculature in coronary disease. Patologia Polska 1994; 45: 17-28. [ Links ]
27. KELM M, SCHAFER S, PREIK M, SCHOEBEL FC, PREIKSTEINHOFF H, STRAUBER BE: Coronary Vasomotion in coronary heart disease-significance of epicardial and microcirculatory vessels from myocardial perfusion. Z Kardiol 1997; 86 (Suppl. 1): 33-41. [ Links ]
28. HEARSE DJ, MAXWELL L, SALDANHA C, GAVIN JB: The myocardial vasculature during ischemia and reperfusion: a target for injury and protection. J Moll Cell Cardiol 1993; 25: 759-800. [ Links ]
29. GAVIN JB, HUMPHREY SM, HERDON PB: The no reflow phenomenon in ischaemic myocardium. Int Rev Expt Pathol 1983; 25: 361-383. [ Links ]
30. KLONER RA, GANOTE CE, JENNINGS RB: The "no reflow" phenomenon after temporary coronary occlusion in the dog. J Clin Invest 1974; 54: 1496-1508. [ Links ]
31. SAGE MD, GAVIN JB: Microvascular function at the margins of early experimental infarcts in isolated rabbit hearts. Heart Vessels 1986; 2: 81-86. [ Links ]
32. MICHAELS AD, GIBSON CM, BARRON HV: Microvascular Dysfunction in Acute Myocardial Infarction: Focus on the Roles of Platelet and Inflammatory Mediators in the No-Reflow Phenomenon. Am J Cardiol 2000; 85: 50B-60B. [ Links ]
33. DREYER W, MICHAEL L, WEST M: Neutrophil accumulation in ischemic canine myocardium: insights into the time course, distribution, and mechanism of localization during early reperfusion. Circulation 1991; 84: 400-11. [ Links ]
34. KLONER RA, GIACOMELLI F, ALKER KJ, MATHEWS R, BELLOWS S: Influx of neutrophils into the walls of large coronary arteries in response to ischemia/ reperfusion. Circulation 1991; 84: 1758-1772. [ Links ]
35. SHERIDAM FM, COLE PG, RAMAGE D: Leucocyte adhesion to the coronary microvasculature during ischemia and reperfusion in an in vivo canine model. Circulation 1996; 93: 1784-1787. [ Links ]
36. ENGLER RL, SCHMITH-SCHONBEIN GW, PAVELEC RS: Leucocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol 1983; 111: 98-111. [ Links ]
37. SIMINIAK T, EGDELL RM, O' GORMAN DJ, DYE JF: Plasma-mediated neutrophil activation during acute myocardial infarction: role of platelet activating factor. Clin Sci 1995; 89: 171-176. [ Links ]
38. KUKIELKA GL, SMITH CW, MANNING AM, YOURKER KA, MICHAEL LH, ENTMAN ML: Induction of interleukin-6 synthesis in the myocardium: potential rle in post-reperfusion inflammatory injury. Circulation 1995; 92: 1866-1875. [ Links ]
39. FRANGOGIANNIS NG, YOURKER KA, ROSSEN RD; GWECHENBERGER M, LINDSEY MH, MENDOZA LH ET AL: Cytoquines and the microcirculation in ischemia and reperfusion. J Mol Cell Cardiol 1998; 30: 2567-2576. [ Links ]
40. ROMSOM JL, HOOK BG, KUNEL SL, ABRAMS GD, SCHORK A, LUCCHESI BR: Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 1983; 67: 1016-1023. [ Links ]
41. YASUDA M, TAKEUCHI K, HIRUMA M, IIDA H, TAHARA A, ITAGANE H ET AL: The complement system in ischemic heart disease. Circulation 1990; 81: 156-163. [ Links ]
42. LANGLOIS PF, GAWRYL MS: Detection of the terminal complement complex in patients plasma following acute myocardial infarction. Atherosclerosis 1988; 70: 95-105. [ Links ]
43. GICLAS PC, PINCKARD RN, OLSON MS: In vivo activation of complement by isolated heart subcellular membranes. J Immunol 1979; 122: 146-151. [ Links ]
44. PINKARD RN, O'ROURKE RA, CRAWFORD MH: Complement localization and mediation of ischemic injury in balloon myocardium. J Clin Invest 1983; 66: 1050-1056. [ Links ]
45. FLETCHER MP, STAHL GL, LONGHURST JC: C5a-induced myocardial ischemia: role for CD18-dependent PMN localization and PMN-platelet interactions. Am J Physiol 1993; 265: H 1750-H 1761. [ Links ]
46. WEYRICH AS, MA XL, LEFER DJ; ALBERTINE KH, LEFER AM: In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J Clin Invest 1993; 91: 2620-2629. [ Links ]
47. SHIMOMURA H, OGAWA H, ARAI H, MORIYAMA Y, TAKAZOE K, HIRAI N ET AL: Serial changes in plasma level of soluble P-selectin in patients with acute myocardial infarction. Am J Cardiol 1998; 81: 397-400. [ Links ]
48. BROWN KK, HENSON PM, MACLOUF J, MOYLE M, ELY JA, WORTHEN GS: Neutrophil-platelet adhesion: relative roles of platelet-P selectin and neutrophil b2 (CD 18) integrins. Am J Respir Cell Moll Biol 1998; 18: 100-110. [ Links ]
49. SHEIKH S, NASH GB: Continuous activation and deactivation of integrin CD11b/CD18 during de novo expression enables rolling neutrophils to immobilize on platelets. Blood 1996; 87: 5040-5050. [ Links ]
50. PEREZ RG, ARAI M, RICHARDSON C, DIPAULA A, SIU C, MATSUMOTO N ET AL: Factors modifying protective effect of anti-CD 18 antibodies on myocardial reperfusion injury in dogs. Am J Physiol 1996; 270: H53-H64. [ Links ]
51. ENGLER RL: Free radical and granulocyte-mediated injury during myocardial ischemia and reperfusion. Am J Cardiol 1989; 63: 19E-23E. [ Links ]
52. KLONER RA: Does reperfusion injury exist in humans? J Am Coll Cardiol 1993; 21: 537-545. [ Links ]
53. ENTMAN ML, YOUKER K, SHOJI T, KUKIELA G, SHAPPEL SB, TAYLOR AA: Neutrophil induced oxidative injury of cardiac myocites: a compartmented system requiring CD 11/CD18-ICAM-1 adherence. J Clin Invest 1992; 90: 1335-1345. [ Links ]
54. PRYZKLENK K, KLONER RA: Superoxide dismutasa plus catalasa improve contractile function in the canine model of the "stunned myocardium". Circ Res 1986; 58: 148-156. [ Links ]
55. HANSEN PR: Role of neutrophils in myocardial ischemia and reperfusion. Circulation 1995; 91: 1872-1885. [ Links ]
56. LUCCHESI BR: Modulation of leucocyte-mediated myocardial reperfusion injury. Ann Rev Physiol 1990; 52: 561-576. [ Links ]
57. DE SCHEERDER IK, VANDEKRAAY AMM, LAMERS JMJ, KOSTER JF, DE JONG JW, SERRUYS PW: Myocardial malondialdehyde and uric acid release after short-lasting coronary occlusions during coronary angioplasty: potential mechanism for free radical generation. Am J Cardiol 1991; 68: 392-395. [ Links ]
58. ROBERTS MJ, YOUNG IS, TROUTON TG, TRIMBLE ER, KHAN MM, WEBB SW ET AL: Transient release of lipid peroxides after coronary artery balloon angioplasty. Lancet 1990; 336: 143-145. [ Links ]
59. DAVIES SW, RANJADAYALAN K, WICKENS DG, DORU-RANDY TL, TIMMIS AD: Lipid peroxidation associated with successful thrombolysis. Lancet 1990; 335: 741-743. [ Links ]
60. FORMAN MB, PERRY JM, WILSON BH; VERANI MS, KAPLAN PR, SHAWL FA, FRESINGER GC: Demonstration of myocardial reperfusion injury in humans: results of a pilot study utilizing acute coronary angioplasty with perfluorochemical in anterior myocardial infarction. J Am Coll Cardiol 1991; 18: 911-918. [ Links ]
61. WALL T, CALIFF R, BLANKENSHIP J: Intravenous Fluosol in the treatment of acute myocardial infarction: results of the Thrombolysis in myocardial infarction 9 Trial. Circulation 1994; 90: 114-120. [ Links ]
62. WERNS SW, BRINKER J, GRUBER J, ROTHBAUM D, HEUSER R, GEORGE B ET AL: A randomized double blind trial of recombinant human superoxide dismutasa (SOD) in patients undergoing PTCA for acute MI. Circulation 1989; 80 (Suppl. II) II-113. [ Links ]
63. FLAHERTY JT, PITT B, GRUBER JW, HEUSER RR, ROTHBAUM D, BURWELL LR, ET AL: Recombinant human superoxide dismutasa (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation 1994; 89: 1982-1991. [ Links ]
64. TOPOL EJ, ELLIS SG, CALIFF RM, GEORGE BS, STUMP DC, BATES ER, ET AL: Combined tissue-type plasminogen activator and prostacyclin therapy for acute myocardial infarction. Thrombolysis and Angioplasty in Myocardial Infarction (TAMI) 4 study Group. J Am Coll Cardiol 1989; 14: 877-884. [ Links ]
65. TOPOL EJ, YADAV JS: Recognition of the importance of embolization in Atherosclerotic Vascular Disease. Circulation 2000; 101: 570-580. [ Links ]
66. ABDELMEGUID AE, TOPOL EJ: The myth of the myocardial "infarctlet" during percutaneous coronary revascularization procedures. Circulation 1996; 94: 3369-3375. [ Links ]
67. KLONER RA, ARIMIE RB, KAY GL, CANNOM D, MATTHEWS R, BHANDARI A, ET AL: Evidence for stunned myocardium in humans: a 2001 update. Coron Artery Dis 2001; 12: 349-356. [ Links ]
68. KLONER RA, PRZYKLENK K, WHITTAKER P: Deleterious effects of oxygen radicals in ischemia/ reperfusion: resolved and unresolved issues. Circulation 1989; 80: 1115-1127. [ Links ]
69. AMBROSIO G, BECKER LC, HUTCHINS GM, WEISMAN HF, WEISFELDT ML: Reduction in experimental infarct size by recombinant human superoxide dismutasa: insights into the pathophysiology of reperfusion injury. Circulation 1986; 74: 1424-1433. [ Links ]
70. BECKER LC, SCHAPER J, JEREMY R, SCHAPER W: Severity of ischemia determines the occurrence of myocardial reperfusion injury. Circulation 1991; 84 (Suppl II): II-254. [ Links ]
71. GANZ W, WATANABE I, KANAMASA K, YANO J, HAN DS, FISHBEIN MD: Does reperfusion extend necrosis? A study in a single territory of myocardial ischemia-half perfused and half no reperfused. Circulation 1990; 82: 1020-1033. [ Links ]
72. KLONER RA, PRZYKLENK K: Experimental infarct size reduction with calcium channel blockers. J Am Coll Cardiol 1991; 18: 876-878. [ Links ]
73. OLAFSON B, FORMAN MB, PUETT DW: Reduction of reperfusion injury in the canine preparation by intracoronary adenosine: importance of the endothelium and the no-reflow phenomenon. Circulation 1987; 76: 1135-1145. [ Links ]
74. DEGRAEFF PA, VAN GILST WH, BEL K, DE LANGEN CDJ, KINGMA JH, WESSELING H: Concentration-dependent protection by captopril against myocardial damage during ischemia and reperfusion in a closed-chest pig model. J Cardiovasc Pharmacol 1987; 9 (Suppl. 2):537-542. [ Links ]
75, NABEL EG, TOPOL EJ, GALEANA A: A randomized placebo-controlled trial of combined early intravenous captopril and recombinant tissue-type plasminogen activator in acute myocardial infarction. J Am Coll Cardiol 1991; 17: 467-473. [ Links ]
76. KLONER RA: Adjuvant pharmacologic therapy for acute myocardial infarction. Hosp Formul 1991; 17: 467-473. [ Links ]
77. THE TIMI STUDY GROUP: Comparison of invasive and conservative strategies after treatment with intravenous tissue plasminogen activator in acute myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27: 335-371. [ Links ]
78. KLONER RA, ALKER K, CAMPBELL C, FIGURES G, EISENHAUER A, HALE S: Does tissue-type plasminogen activator have direct beneficial effects on the myocardium independent of its ability to lyse intracoronary thrombi? Circulation 1989; 79: 1125-1136. [ Links ]
79. AMBROSIO G, WEISMAN HF, MANSINI JA, BECKER LC: Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation 1989; 80: 1846-1861. [ Links ]
80. KU DD: Coronary vascular reactivity after myocardial ischemia. Science 1982; 218: 576-578. [ Links ]
81. VAN BENTHUYSEN KM, MCMURTY IF, HORWITZ LD: Reperfusion after coronary occlusion in dogs impairs endothelium dependent relaxation to acetylcoline and augments contractile reactivity in vitro. J Clin Invest 1987; 79: 265-274. [ Links ]
82. QUILLEN JE, SELKE FW, BROOKS LA, HARRISON DG: Ischemia-reperfusion impairs endothelium- dependent relaxation of coronary microvessels but does not affect large arteries. Circulation 1990; 82: 586-594. [ Links ]
83. TSAO PS, AOKI N, LEFER DJ, JOHNSON G, LEFER AM: Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation 1990; 82: 1402-1412. [ Links ]
84. SCHOEFER J, MONTZ R, MATHEY DG: Scintigraphic evidence of the "no reflow" phenomenon in humans beigns after coronary thrombolysis. J Am Coll Cardiol 1985; 5: 593-598. [ Links ]
85. ITO H, TOMOOKA T, SAKI N: Lack of myocardial perfusion immediately after successful thrombolysis. A predictor of poor recovery of left ventricular function in anterior myocardial infarction. Circulation 1992; 85: 1699-1705. [ Links ]
86. KLONER RA, ALKER KJ: The effect of streptokinase on intramyocardial hemorrhage infarct size, and the no-reflow phenomenon during coronary reperfusion. Circulation 1984; 70: 513-521. [ Links ]
87. ROBERTS CS, SCHOEN FJ, KLONER RA: Effect of coronary reperfusion on myocardial hemorrhage and infarct healing. Am J Cardiol 1983; 52: 610-614. [ Links ]
88. FISHBEIN MC, RIT J, LANDO V, KANMATSUSE R, MERCIER JC, GANZ W: The relationship of vascular injury and myocardial hemorrhage to necrosis after reperfusion. Circulation 1980; 62: 1274-1279. [ Links ]
89. GERTZ SD, KALAN JM, KRAGEL AN, ROBERTS WC, BRAUNWALD E: The TIMI Investigators. Cardiac morphologic findings in patients with acute myocardial infarction treated with recombinant tissue plasminogen activator. Am J Cardiol 1990; 65: 953-961. [ Links ]
90. BRAUNWALD E, KLONER RA: The stunned myocardium: prolonged, post-ischemic ventricular dysfunction. Circulation 1982; 66:1146-1149. [ Links ]
91. BOLLI R, JEROUDI MO, PATEL BS: Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial "stunning" is a manifestation of reperfusion injury. Circ Res 1989; 65: 607-622. [ Links ]
92. TOUCHTONE DA, BELLER GA, NYGAARD TW, TEDESCO C, KAUL S: Effects of successful intravenous reperfusion therapy on regional myocardial function and geometry in humans: a tomographic assessment using two-dimensional echocardiography. J Am Coll Cardiol 1989; 13: 1506-1513. [ Links ]
93. SHEEHAN FH, DOERR R, SCHMITH WG: Early recovery of left ventricular function after thrombolytic therapy for acute myocardial infarction: an important determinant of survival. J Am Coll Cardiol 1988; 12: 289-300. [ Links ]
94. PFISTERER M, ZUBER M, WENZEL R, BURKART F: Prolonged myocardial stunning after thrombolysis: can left ventricular function be assessed definitely at hospital discharge? Eur Heart J 1991; 12: 214-217. [ Links ]
95. NIXON JV, BROWN CN, SMITHERMAN TC: Identification of transient and persistent segmental wall motion abnormalities in patients with unstable angina by two dimmensional echocardiography. Circulation 1982; 65: 1497-1503. [ Links ]
96. WILSON IC, GARDNER TJ: Stunned myocardium in Cardiac Surgery. In: Kloner RA, Przyklenk K Ed. The stunned Myocardium: properties, mechanisms, and clinical manifestations. New York: Marcel Dekker, 1993: 379-399. [ Links ]
97. WIJNS W, SERRUYS PW, SLAGER CJ: Effects of coronary occlusion during percutaneous transluminal angioplasty in humans on left ventricular chamber stiffness and regional pressure radius relations. J Am Coll Cardiol 1986; 7: 455-463. [ Links ]
98. KLONNER RA, ALLEN J, COX TA, ZHENG Y, RUÍZ CF: Stunned left ventricular myocardium following exercise treadmill testing in coronary artery disease. Am J Cardiol 1991; 68: 629-634. [ Links ]
99. ARNOLD JMO, BRAUNWALD E, SANDOR T, KLONER RA: Inotropic stimulation of reperfused myocardium: effects on infarct size and myocardial function. J Am Coll Cardiol 1985; 6: 1026-1034. [ Links ]
100. SATLER LF, KENT KM, FOX LM: The assessment of contractile reserve after thrombolytic therapy for acute myocardial infarction. Am Heart J 1986; 111: 821-825. [ Links ]
101. MANNING AS, HEARSE DJ: Reperfusion-induced arrhythmias: mechanisms and prevention. J Moll Cell Cardiol 1984; 16: 497-518. [ Links ]
102. HALE SL, LANGE R, ALKER KJ, KLONER RA: Correlates of reperfusion ventricular fibrillation in dogs. Am J Cardiol 1984; 53: 1397-1400. [ Links ]
103. HAGAR JM, HALE SL, KLONER RA: Effect of pre-conditioning ischemia on reperfusion arrhythmias after coronary artery occlusion and reperfusion in the rat. Circ Res 1991; 68: 61-68. [ Links ]
104. HACKETT D, MCKENNA W, DAVIES G, MASERI A, LOWE T, ABER V: Reperfusion arrhythmias are rare during acute myocardial infarction and thrombolysis in man. Int J Cardiol 1990; 29: 205-213. [ Links ]
105. BURNEY RE, WALSH D, KAPLAN LR, FRASER S, TUNG B, OVERMYER J: Reperfusion arrhythmia: myth or reality. Ann Emerg Med 1989; 18: 240-243. [ Links ]
106. DAVIES MJ, THOMAS AC, KNAPMAN PA, HANGARTNER JR: Intramyocardial platelet aggregation in patients with unstable angina suffering sudden ischemic cardiac death. Circulation 1986; 73: 418-427. [ Links ]
107. FRINK RJ, ROONEY PA JR, TROWBRIDGE JO, ROSE PJ: Coronary thrombosis and platelet/fibrin microemboli in death associated with acute myocardial infarction. Br Heart J 1988; 59: 196-200. [ Links ]
108. FARB A, TANG AL, BURKE AP, SESSUMS L, LIANG Y, VIRMANI R: Sudden coronary death: frequency of active coronary lesions, inactive coronary lesions, and myocardial infarction. Circulation 1995; 92: 1701-1709. [ Links ]
109. FRINK RJ, OSTRACH LH, ROONEY PA, ROSE J: Coronary thrombosis, ulcerated atheroesclerotic plaques and platelet/fibrin microemboli in patients dying with acute coronary disease: a large autopsy study. J Clin Invest 1990; 2: 199-210. [ Links ]
110. FALK E, SHAH PK, FUSTER V: Coronary plaque disruption. Circulation 1995; 92: 657-671. [ Links ]
111. GASSLER JP, TOPOL EJ: Reperfusion Revisited: beyond TIMI 3 flow. Clin Cardiol 1999; 22: IV 20-IV 29. [ Links ]
112. DOUGLAS JS JR: Percutaneous intervention in patients with prior coronary bypass surgery. In: Topol EJ Ed. Textbook of Interventional Cardiology 3er. Ed. Philadelpia Pa: WB Saunders, 1999: 297-316. [ Links ]
113. EL-MARAGHI N, GENTON E: The relevance of platelet and fibrin thromboembolism of the coronary microcirculation, with special reference to sudden cardiac death. Circulation 1980; 62: 936-944. [ Links ]
114. FOLTS JD, CROWELL EB, ROWE GG: Platelet aggregation in partially obstructed vessels and its elimination with aspirin. Circulation 1976; 54: 365-370. [ Links ]
115. HIRSH PD, HILLIS LD, CAMPBELL WB, FIRTH BG, WILLERSON JT: Release of prostaglandins and thromboxane into the coronary circulation in patients with ischemic heart disease. N Engl J Med 1981; 304: 685-691. [ Links ]
116. EIDT JF, ALLISON P, NOBLE S, ASHTON J, GODINO P, MCNATT J, ET AL: Thrombin is an important mediator of platelet aggregation in stenosed canine coronary arteries with endothelial injury. J Clin Invest 1989; 84: 18-27. [ Links ]
117. YAO S-K, OBER JC, MCNATT J, BENEDICT CR, ROSOLOWSKY M, ANDERSON HV, ET AL: ADP plays an important role in mediating platelet aggregation and cyclic flow variations in vivo in stenosed and endothelium injured canine coronary arteries. Circ Res 1992; 70: 39-48. [ Links ]
118. DAVIES MJ: The composition of coronary-artery plaques. N Engl J Med 1997; 336: 1312-1313. [ Links ]
119. HEESCHEN C, VAN DEN BRAND MJ, HAMM CW, SIMMONS ML, FOR THE CAPTURE INVESTIGATORS: Angiographic Findings in Patients with Refractory Unstable Angina According to Troponin T Status. Circulation 1999; 104: 1509-1514. [ Links ]
120. ZAHI F, FUSTER V, FALLON JT, JAYASUNDERA T, WORTHLEY SG, HELFT G, ET AL: Noninvasive In Vivo Human Coronary Artery Lumen and Wall Imaging Using Black-Blood Magnetic Resonance Imaging. Circulation 2000; 102: 505-510. [ Links ]
121. TOPOL EJ, LEYA F, PINKERTON CA, WITHLOW PL, HOFLING B, SIMONTON CA, ET AL FOR THE CAVEAT STUDY GROUP: A comparison of coronary angioplasty with directional atherectomy in patients with coronary artery disease. N Engl J Med 1993; 329: 221-227. [ Links ]
122. HOLMES DR,TOPOL EJ, CALIFF RM, LEYA F, BERBERG PB, TALLEY JD, ET AL: For the CAVEAT-II Investigators: A multicenter, randomized trial of coronary angioplasty versus directional atherectomy for patients with saphenous vein graft lesions. Circulation 1995; 91: 1966-1974. [ Links ]
123. EGUCHI H, IKEDA K, MUROHARA T, YASUKAWA H, HARAMAKI N, SAKISAKA S, IMAIZUMI K: Endothelial injuries of coronary arteries distal to thrombotic sites: role of adhesive interaction between endothelial P-selectin and leukocyte sialyl Lewis X. Circ Res 1999; 84: 525-535. [ Links ]
124. LASTER SB, OHNISHI Y, SAFFITZ JE: Effects of reperfusion on ischemic right ventricular dysfunction: Disparate mechanisms of benefit related to duration of ischemia. Circulation 1994; 90: 1398-1409. [ Links ]
125. THE GLOBAL USE OF STRATEGIES TO OPEN OCCLUDED CORONARY ARTERIES IN ACUTE CORONARY SYNDROMES (GUSTO IIB) ANGIOPLASTY SUBSTUDY INVESTIGATORS. A clinical trial comparing primary coronary angioplasty with tissue plasminogen activator for acute myocardial infarction. N Engl J Med 1997; 336: 1621-1628. [ Links ]
126. GIBSON CM, CANNON CP, PIANA RN, BREALL JA, SHARAF B, FLATLEY M, ET AL: Angiographic predictors of reocclusion after thrombolysis in Myocardial Infarction (TIMI) 4 trial. J Am Coll Cardiol 1995; 25: 582-589. [ Links ]
127. TAN WA, MOLITERNO DJ: TIMI flow and surrogate end points: What you see is not always what you get. Am Heart J 1998; 136: 570-573. [ Links ]
128. GIBSON CM, CANNON CP, DALEY WL, DODGE JT, ALEXANDER B, MARBLE SJ, ET AL: The TIMI frame count: a quantitative method of assessing coronary artery flow. Circulation 1996; 93: 879-888. [ Links ]
129. GIBSON CM, GOEL M, DOTANI I, RIZO M, MCLEAN C, MARTIN NE ET AL: The post-PTCA TIMI frame count and mortality in RESTORE. Circulation 1996; 94 (suppl I): 1-85. [ Links ]
130. GIBSON CM, CANNON CP, MURPHY SA, RYAN KA, MESLEY R, MARBLE SJ, ET AL: Relationship of TIMI myocardial perfusion grade to mortality after administration of thrombolytic drugs. Circulation 2000; 101: 125-130. [ Links ]
131. WU KC, ZERHOUNI EA, JUDD RM, LUGO-OLIVIERI CH, BAROUCH LA, ET AL: Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 1998; 97: 765-772. [ Links ]
132. VALLEJO E, PEÑA-DUQUE MA, NOROÑO O, BANHAYASHI E, GASPAR J, VILLAVICENCIO R, MARTÍNEZ RMA: Fenómeno de no-reflujo: incidencia y características clínicas en una serie de casos. Arch Inst Cardiol Méx 1998; 68: 247-252. [ Links ]
133. CALIFF RM, ABELMEGUID A, KUNTZ R, POPMA JJ, TARDIFF BE, CRENSHAW B, ET AL: Myonecrosis after revascularization procedures. J Am Coll Cardiol 1998; 31: 241-251. [ Links ]
134. ADGEY AAJ, MATHEW TP, HARBINSON MT: Periprocedural creatine kinase-MB elevations: long-term impact and clinical implications. Clin Cardiol 1999; 22: 257-265. [ Links ]
135. EPISTENT INVESTIGATORS: Randomized controlled trial to assess safety of coronary stenting with use of abciximab. Lancet 1998; 352: 85-90. [ Links ]
136. JOHANSEN O, BREKKE M, STROMMME JH, VALEN V, SELJEFLOT I, SKJAEGGESTAD O, ARNESEN H: Myocardial damage during percutaneous transluminal coronary angioplasty as evidenced by troponin T measurements. Eur Heart J 1998; 19: 112-117. [ Links ]
137. REIMERS B, LACHIN M, CACCIOVILLANI L, SECCHIERO S, RAMONDO A, ISABELLA G, ET AL: Troponin T, creatine kinasa MB mass and creatine kinasa isoform ratio in the detection of myocardial damage during non-surgical coronary revascularization. Int J Cardiol 1997; 60: 7-13. [ Links ]
138. HAMM CW, HEESCHEN C, GOLDMAN B, VAHANIAN A, ADGEY J, MIGUEL CM, ET AL FOR THE CAPTURE INVESTIGATORS. Benefit of abciximab in patients with refractory unstable angina in relation to troponin T levels. N Engl J Med 1999; 340: 1623-1629. [ Links ]
139. GASPARONE A, CREA F, VERSACI F, TOMAI F, PELLEGRINO A, CHIARIELLO L, GIOFFRE PA: Predictive value of C-reactive protein after successful coronary artery stenting in patients with stable angina. Am J Cardiol 1998; 82: 515-518. [ Links ]
140. MORROW DA, RIFAI N, ANTMAN EM, WIENER DL, MCCABE CH, CANNON CP, BRAUNWALD E: C-reactive protein is a potent predictor of mortality independently of and in combination with troponin T in acute coronary syndromes: a TIMI 11-A substudy. J Am Coll Cardiol 1998; 31: 1460-1465. [ Links ]
141. FOX KAA: ACUTE CORONARY SYNDROMES: presentation-clinical spectrum and management. Heart 2000; 84: 93-100. [ Links ]
142. FURMAN MI,DAUERMAN HL, GOLDBERG RJ, YARZBESKI J, LESSARD D, GORE JM: Twenty-Two Year (1975 to 1997) Trens in the Incidence, In- ospital and long-term Case fatality Rates From Initial Q-wave and non-Q-wave Myocardial Infarction: A Multi-hospital, Community-Wide Perspective. J Am Coll Cardiol 2001; 37: 1571-1580. [ Links ]
143. BOERSMA E, HARRINGTON RA, MOLITERNO DJ, WHITE H, TEROUX P, VAN DE WERF F, ET AL: Plate- let glycoprotein IIb/III a inhibitors in acute coronary syndromes: a meta-analysis of all major randomized trials. Lancet 2002; 359: 189-198. [ Links ]
144. BERTRAND ME, SIMMONS ML, FOX KAA: Management of acute coronary syndromes: acute coronary syndromes without persistent ST segment elevation: recommendation of the Task Force of the European Society of Cardiology. Eur Heart J 2000; 21: 1406-1432. [ Links ]
145. STONE GW, BRODIE BR, GRIFFIN JJ, MORICE MC, CONSTANTINI C, OVERLIE PA, ET AL ON BEHALF OF THE PRIMARY ANGIOPLASTY IN MYOCARDIAL INFARCTION (PAMI) INVESTIGATORS: Improved short-term outcomes of primary coronary stenting compared to primary balloon angioplasty in acute myocardial infarction at experienced centers: the PAMI study group experience. J Intervent Cardiol 1999; 12: 101-108. [ Links ]
146. GRINES CL,COX DA, STONE GW: Coronary angioplasty with or without stent implantation for acute myocardial infarction. N Engl J Med 1999; 341: 1949-1956. [ Links ]
147. BRODIE BR, STUCKEY TD: Mechanical reperfusion therapy for acute myocardial Infarction: stent PAMI, ADMIRAL, CADILLAC and beyond. Heart 2002; 87: 191-192. [ Links ]
148. THE GUSTO V INVESTIGATORS: Reperfusion therapy for acute myocardial infarction with fibrinolytic therapy or combination reduced fibrinolytic therapy and platelet glycoprotein IIb/III a inhibition: The GUSTO V randomized trial. Lancet 2001; 357: 1905-1914. [ Links ]
149. STONE GW, GRINES CL, COX DA, GARCÍA E, TCHENG JE, GRIFFIN JJ ET AL: For the controlled abciximab and device investigation to lower late angioplasty complications (CADILLAC) investigators: Comparison of angioplasty with stenting, with or without abciximab, in acute myocardial infarction. N Engl J Med 2002; 346: 957-966. [ Links ]
150. THE ASSESSMENT OF THE SAFETY AND EFFICACY OF A NEW THROMBOLYTIC REGIMEN (ASSENT)-3 INVESTIGATORS: Efficacy and safety of tenecteplase in combination with enosaparin, abciximab, or unfractionated heparin: The ASSENT-3 randomized trial in acute myocardial infarction. Lancet 2001; 358: 605-613. [ Links ]
151. MONTALESCOT G, BARRAGAN P, WITTENBEG O, ECOLLAN P, ELHADAD S, VILLAIN P ET AL: For the ADMIRAL Investigators: Platelet glycoprotein IIb/III a inhibition with coronary stenting for acute myocardial infarction. N Engl J Med 2001; 344: 1895-1903. [ Links ]
152. NEUMAN JF, BLASINI R, SCHMITT C, ECKHARD A, DIRSCHINGER J, GAWAZ M ET AL: Effect of glycoprotein IIb/III a receptor blockade on recovery of coronary flow and left ventricular function after the placement of coronary-artery stents in acute myocardial infarction. Circulation 1998; 98: 2695-2701. [ Links ]
153. FRANGOGIANNIS NG, SMITH WC, ENTMAN ML: The inflammatory respose in myocardial infarction. Cardiovasc Res 2002; 53: 31-47. [ Links ]
154. ENTMAN ML, SMITH CW: Postreperfusion inflammation: a model for reaction to injury in cardiovascular disease. Cardiovasc Res 1994; 9: 1301-1311. [ Links ]
155. LIBBY P, MAROKO PR, BLOOR CM, SOBEL BE, BRAUNWALD E: Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest 1973; 3: 599-607. [ Links ]
156. ROBERTS R, DEMELLO V, SOBEL BE: Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation 1976; 1: 204-206. [ Links ]
157. KLONER RA, FISHBEIN MC, LEW H, MAROKO PR, BRAUNWALD E: Mummification of the infarcted myocardium by high dose corticosteroids. Circulation 1978; 1: 56-63. [ Links ]
158. HILL JH, WARD PA: The phlogistic role of C3 leukotactic fragment in myocardial infarcts of rats. J Exp Med 1971; 1: 885-890. [ Links ]
159. PINCKARD RN, OLSON MS, GICLAS: Consumption of classical complement components by heart subcellular membranes in vitro and in patients after acute myocardial infarction. J Clin Invest 1975; 3: 740-750. [ Links ]
160. GILLUM RF, INGRAM DD, MAKUC DM: White blood cell count, coronary heart disease, and death: the NHANES 1 epidemiologic follow-up study. Am Heart J 1993; 125: 855-863. [ Links ]
161. FRIEDMAN GD, KLASTKY AL, SIEGELAUB AB: The leukocyte count as a predictor of myocardial infarction. N Engl J Med 1974; 290: 1275-1278. [ Links ]
162. FRANGI D, GARDINALI M, CONCIATO L, CAFARO C, POZZONI L, AGOSTINO A: Abrupt complement activation and transient neutropenia in patients with acute myocardial infarction treated with streptokinase. Circulation 1994; 89: 76-80. [ Links ]
163. PIETILA KO, HARMOINEN AP, JOKINITTY J, PASTERNACK AI: Serum C-reactive protein concentration in acute myocardial infarction and its relationship to mortality during 24 months of follow-up in patients under thrombolytic treatment. Eur Heart J 1996; 17: 1345-1349. [ Links ]
164. MEIJER CJ, HACK CE: C reactive protein colocalizes with complement in human hearts during acute myocardial infarction. Circulation 1997; 95: 97-103. [ Links ]
165. RIDKER PM, RIFAI N, PFEFFER MA, SACKS FM, MOYE LA, GOLDMAN S, ET AL: Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average colesterol levels. Circulation 1998; 98: 839-844. [ Links ]
166. JUÁREZ UH, LASSES LA, MARTÍN RP, LUNA JG, LÓPEZ MCR, CHUQUIURE EV, ET AL: Utilidad de la determinación cualitativa rápida de troponina T, fracción MB de creatinina fosfocinasa y mioglobina en los síndromes isquémicos coronarios agudos. Arch Inst Cardiol Méx 1998; 68: 473-481. [ Links ]
167. SARAN RK, BEEN M, FURNISS SS, HAWKINS T, REID DS: Reduction in ST segment elevation after thrombolysis predicts either coronary reperfusion or preservation of left ventricular function. Br Heart J 1990; 64: 113-117. [ Links ]
168. BARBASH GI,ROTH A, HOD H, MILLER HI, RATH S, HARZAHAV Y, MODAN M, ET AL: Rapid resolution of ST elevation and prediction of clinical outcome in patients undergoing thrombolysis with alteplase (recombinant tissue-type plasminogen activator): results of the Israeli Study of Early Intervention in Myocardial Infarction. Br Heart J. 1990; 64: 241-247. [ Links ]
169. MAURI F, MAGGIONI AP, FRANZOSI MG, VITA C, SANTORO E, SANTORO L ET AL: A simple electrocardiographic predictor of the outcome of patients with acute myocardial infarction treated with a thrombolytic agent. A GISSI-2 derived analysis. J Am Coll Cardiol 1994; 24: 600-607. [ Links ]
170. DISSMANN R, SCHRÖEDER R, BUSSE U, APPEL M, BRÜGGEMANN T, JERECZEK M, LINDERER T: Early assessment of outcome by segment analysis after thrombolytic therapy in acute myocardial infarction. Am Heart J 1994; 128: 851-857. [ Links ]
171. SCHROEDER RI, WEGSCHEIDER K, SCHROEDER K, DISSMANN R, MEYER- SABELLEK W: Extent of early ST segment elevation resolution: A strong predictor of outcome in patients with acute myocardial infarction and a sensitive measure to compare thrombolytic regimens. A substudy of the International Joint Efficacy Comparison of Thrombolytics (INJECT) Trial. J Am Coll Cardiol 1995; 26: 1657-1664. [ Links ]
172. NAKAMURA M, TSUNODA T, WAKATSUKI T, UI K, DEGAWA T, YABUKI S, YAMAGUCHI T: Distal coronary flow velocity immediately after direct angioplasty for acute myocardial infarction. Am Heart J 1996; 132: 251-257. [ Links ]
173. IWAKURA K, ITO H, TAKUCHI S, TANIYAMA Y, NAKATSUCHI Y, NEGORO S, ET AL: Alteration in the coronary blood flow velocity pattern in patients with no reflow and reperfused acute myocardial infarction. Circulation 1996; 94: 1269-1275. [ Links ]
174. VANHAECKE J, FLAMENG W, BORGERS M, JANG IK, VAN DE WERF F, DEGEEST H: Evidence for decreased coronary flow reserve in viable postischemic myocardium. Cir Res 1990; 67: 1200-1210. [ Links ]
175. BROCHET E, CZITROM D, KARILA-COHEN D, SEKNADJI P, FARAGGI M, BENAMER H, ET AL: Early changes in myocardial perfusion patterns after myocardial infarction: relation with contractile reserve and functional recovery. J Am Coll Cardiol 1998; 32: 2011-1017. [ Links ]
176. SAKUMA T,HAYASHI Y, SUMII K, IMAZU M, YAMAKIDO M: Prediction of short-and intermediate-term prognoses of patients with acute myocardial infarction using myocardial contrast echocardiography one day after recanalization. J Am Coll Cardiol 1998; 32: 890-897. [ Links ]
177. KONDO M, NAKANO A, SAITO D, SHIMONO Y: Assessment of "microvascular no-reflow phenomenon" using technectium-99 m macroaggregated albumin scintigraphy in patients with acute myocardial infarction. J Am Coll Cardiol 1998; 32: 898-903. [ Links ]
178. MAES A, VAN DER WERF, NUYTS J, BORMANS G, DESMET W, MORTELMANS L: Impaired myocardial tissue perfusion early after successful thrombolysis: impact on myocardial flow, metabolism and function at late follow-up. Circulation 1995; 92: 2070-2078. [ Links ]
179. MATSUMARA K, JEREMY RW, SCHAPER J, BECKER LC: Progression of myocardial necrosis during reperfusion of ischemic myocardium. Circulation 1998; 97: 795-804. [ Links ]
180. SAEED M, WENDLAND M, TAKEHARA Y, MASUI T, HIGGINS CB: Reperfusion and irreversible myocardial injury: identification with a nonionic MR imaging contrast medium. Radiology 1992; 182: 675-683. [ Links ]
181. LIMA JAC, JUDD RM, BAZILE A, SCHULMAN SP, ATALAR E, ZERHOUNI EA: Regional heterogeneity of human myocardial infarcts demonstrated by contrast enhanced MRI. Potential mechanisms. Circulation 1995; 92: 1117-1125. [ Links ]
182. SAED M, VANDIJKE CF, WENDLAND MF, ROSENAU W, HIGGINS CB, BRASH RC: Histologic confirmation of microvascular hyperpermeability to macromolecular MR contrast medium in reperfused myocardial infarction. J Magn Reson Imaging 1998; 8: 561-567. [ Links ]
183. ITO H, OKAMURA A, IWAKURA K, MASUYAMA T, HORI M, TAKIUCHI S, ET AL: Myocardial perfusion patterns related to thrombolysis in myocardial infarction perfusion grades after coronary angioplasty in patients with acute anterior myocardial infarction. Circulation 1996; 93: 1993-1999. [ Links ]
184. AGATI L, VOCI P, BILOTTA F, LUONGO R, AUTORE C, PENCO M, ET AL: Influence of residual perfusion within the infarct zone on the natural history of left ventricular dysfunction after acute myocardial infarction: a myocardial contrast echocardiography study. J Am Coll Cardiol 1994; 34: 1336-1342. [ Links ]
185. SAEIAN K, RHYNE TL, SAGAR KB: Ultrasonic tissue characterization for diagnosis of acute myocardial infarction in the coronary care unit. Am J Cardiol 1994; 74: 1211-1215. [ Links ]
186. LIN LC, WU CC, HO YL: Ultrasonic tissue characterization for coronary care unit patients with acute myocardial infarction. Ultrasound Med Biol 1998; 24: 187-196. [ Links ]
187. TAKIUCHI S, ITO H, IWAKURA K: Ultrasonic tissue characterization predicts myocardial viability in early stage of reperfused acute myocardial infarction. Circulation 1998; 97: 350-362. [ Links ]
188. LEISTAD E, OHMORI K, PETERSON TA, CHRISTENSEN G, DEMARIA AN: Quantitative assessment of myocardial perfusion during graded coronary artery stenosis by intravenous myocardial contrast echocardiography. J Am Coll Cardiol 2001; 37: 624-631. [ Links ]
189. HANCOCK JE, COOKE JC, CHIN DT, MONAGHAM ML: Determination of successful reperfusion after thrombolysis for acute myocardial infarction. A noninvasive method using ultrasonic tissue characterization that can be applied clinically. Circulation 2002; 105: 157-161. [ Links ]
190. ANTMAN EM, GIUGLIANO RP, GIBSON GM ET AL: For the TIMI 14 Investigators. abciximab facilitates the rate and extent of thrombolysis: results of the thrombolysis in myocardial infarction (TIMI) 14 trial. Circulation 1999; 99: 2720-2732. [ Links ]
191. MIZUMURA T, NITHIPATIKOM K, GROSS GL: Effects of nicorandil and glyceryl trinitrate on infarct size, adenosine realize, and neutrophil infiltration in the dog. Cardiovasc Res 1995; 29: 482-489. [ Links ]
192. SAKATA Y, KODAMA K, LIM YJ, ISHIKURA F, HIRAYAMA A, KITAKAZE M ET AL: Salutary effect of adjuntive intracoronary nicorandil administration on restoration of myocardial blood flow and functional improvement in patients with acute myocardial infarction. Am Heart 1997; 133: 616-621. [ Links ]
193. GALIÉ N, GUARNIERI C, USSIA GP, ZIMARINO M, TAINI AM, PARLANGELI R, ET AL: Limitation of myocardial infarct size by nicorandil after sustained ischemia in pigs. J Cardiovasc Pharmacol 1995; 26: 477-484. [ Links ]
194. PIEPER GM, GROSS GL: Anti-free-radical and neutrophil-modulating properties of the nitrovasodilator, nicorandil. Cardiovasc Drugs Ther 1992; 6: 225-232. [ Links ]
195. MASAZUMI A, LEFER DJ, SO T, DIPAULA A, AVERSANO T, BECKER LC: An anti-CD18 antibody limits infarct size and preserves left ventricular function in dogs with ischemia and 48-hour reperfusion. J Am Coll Cardiol 1996; 27: 1278-1285. [ Links ]
196. MAHAFFEY KW, PUMA JA, BARBAGELATA A, DICARLI MF, LEESAR MA, BROWE KF ET AL: For the AMISTAD Investigators: Adenosine as an adjunt to thrombolytic therapy for acute myocardial infarction: Results of a multicenter, randomized, placebo-controlled trial: The acute miocardial infarction study of adenosine (AMISTAD) trial. J Am Coll Cardiol 1999; 34: 1711-1720. [ Links ]
197. ROSS AM, MOLHOEK P, LUNDERGAN C, KNUDTSON M, DRAOUI Y. REGALADO L RN ET AL: Randomized comparison of enoxaparin, a low-molecular-weight heparin, with unfarctionated heparin adjuntive to recombinant tissue plasminogen activator thrombolysis and aspirin: second trial of heparin and aspirin reperfusion therapy (HEART II). Circulation 2001; 104:648-652. [ Links ]
198. ANTMAN EM, LOUWERENBURG HW, BAARS HF, WESDORP JCL, HAMER B, BASSAND JP ET AL: For the ENTIRE-TIMI 23 Investigators: Enoxaparin as adjunctive antithrombin therapy for ST-Elevation myocardial infarction: Results of the ENTIRE-Thrombolysis in Myocardial Infarction (TIMI) 23 trial. Circulation 2002;104: 1642-1648. [ Links ]
199. COUSSEMENT PK, BASSAND JP, CONVENS C, VROLIX M, BOLAND J, GROLLIER G, ET AL: FOR THE PENTALYSE STUDY: A synthetic factor-Xa inhibitor (ORG31540/sr9017A) as an adjunt to fibrinolysis in acute myocardial infarction. The PENTALYSE study. Eur Heart J 2001; 22: 1716-1724. [ Links ]
200. ROGERS WJ,CANTO JG, LAMBREW CT, TIEFENBRUN AJ, KINKAID B,SHOULTZ DA, ET AL: For the Investigators in the National Registry of Myocardial Infarction 1, 2 and 3: temporal Trends in the Treatment of over 1.5 Million Patients with Myocardial Infarction in the U.S from 1990 throught 1999. J Am Coll Cardiol 2000; 36: 2056-2063. [ Links ]
201. TOPOL EJ, CALIFF RM, GEORGE BS, KEREIAKES DJ, ABOOTTSMITH CW, CANDELA RJ ET AL: A randomized trial of immediate vs delayed elective angioplasty after intravenous tissue plasminogen activator in acute myocardial infarction. N Engl J Med 1987; 317: 581-588. [ Links ]
202. SIMMOONS ML, BETRIUA A, COL J, VON ESSEN R, LUBSEN J, MICHEL PL ET AL: Thrombolysis with tissue plasminogen activator in acute myocardial infarction: no additional benefits from immediate percutaneous coronary angioplasty. Lancet 1988; 1: 197-202. [ Links ]
203. THE TIMI STUDY GROUP: Comparison of invasive and conservative strategies after treatment with intravenous tissue plasminogen activator in acute myocardial infarction: results of the thrombolysis in myocardial infarction (TIMI) Phase II Trial. N Engl J Med 1989; 320: 618-627. [ Links ]
204. O'NEIL WW, WEINTRAUB R, GRINES CL, MEANY B, BRODIE BR, FRIEDMAN HZ, ET AL: A prospective, placebo-controlled, randomized trial of intravenous streptokinase and angioplasty vs alone angioplasty therapy of acute myocardial infarction. Circulation 1992; 86: 1710-1717. [ Links ]
205. ANTMAN EM, GIUGLIANO RP, MCCABE CH,GIBSON M, ADGEY AJJ, GHALI M, ET AL: Abciximab (Re-oPro) potentiates thrombolysis in ST elevation myocardial infarction: results of TIMI 14 Trial (Abst). J Am Coll Cardiol 1998; 319( Suppl A ): 191 A. [ Links ]
206. HERRMANN HC, MOLITERNO DJ, OHMAN EM, STEBBINS AL, BODE C, BETRIU A, ET AL: Facilitation of early percutaneous coronary intervention after re-teplase with or without abciximab in acute myocardial infarction: results from the SPEED (Gusto-4 Pilot) trial. J Am Coll Cardiol 2000; 36: 1989-1996. [ Links ]
207. BRODIE BR, STUCKEY TD, HANSEN C, MUNCY D: Benefit of coronary reperfusion before intervention on outcomes after primary angioplasty for acute myocardial infarction. Am J Cardiol 2000; 85: 13-18. [ Links ]
208. VARGAS BJ, PEÑA DM, ROLDAN FJ, ROMERO AC, ESPÍNDOLA ZN, FÉREZ SS: Detection of right ventricular myocardial perfusion and contractil reserve by contrast echocardiography and low dose dobutamine in myocardial infarction after successful right coronary angioplasty. Arch Cardiol Méx 2002; 72: 49-52. [ Links ]
209. LUPI-HERRERA E, LASSES LA, COSIO AJ, CHUQUIURE VE, MARTÍNEZ SC, ORTÍZ P, ET AL: Acute right ventricular infarction: clinical spectrum, results of reperfusion therapy and short-term prognosis. Coron Artery Dis 2002;13: 57-64. [ Links ]

