











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink“El límite de las aplicaciones derivadas de la edición genética con las herramientas CRISPR está en la imaginación de los investigadores”
Lluís Montoliu, 2019
Introducción
Breve historia de las CRISPR y su función como sistema inmune en las bacterias
Los sistemas CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) y sus proteínas Cas (CRISPR Associated Proteins) son un grupo de genes que actúan de manera simultánea para identificar y eliminar las secuencias de DNA invasoras, funcionando como una memoria de infecciones previas en la mayoría de los procariontes (bacterias, ~50%; arqueas, ~87%) (Makarova et al., 2015; Zhang, Wu, Wu, Chang & Liu, 2021). Estas secuencias fueron descritas por primera vez en 1987 por Ishino y colaboradores como una serie de repeticiones interespaciadas de DNA parcialmente palindrómicas (del griego palin dromein, refiriéndose a una frase que se lee igual hacia adelante que hacia atrás), en la región 3’ río abajo del gen responsable de la conversión de la isoenzima fosfatasa alcalina (iap) de Escherichia coli (Ishino, Shinagawa, Makino, Amemura & Nakatura, 1987). Posteriormente, otros grupos de investigación reportaron secuencias palindrómicas similares de ~29 nucleótidos espaciadas de manera repetida por otros ~32 nucleótidos en especies de bacterias (Mycobacterium tuberculosis) y arqueas (Haloferax mediterranei) (Groenen, Bunschoten, Van Soolingen & van Embden, 1993; Mojica, Juez & Rodríguez-Valera, 1993). Los avances en la secuenciación de genomas en los años 90 hicieron posible encontrar más secuencias de otras especies en los siguientes años. Aunque su estructura y función no estaban del todo dilucidadas, fueron utilizadas en la tipificación de bacterias (Barrangou & Horvath, 2017). En este periodo surgieron distintos acrónimos para nombrarlas como: repeticiones variables directas (DVR), repeticiones en tándem (TREP), secuencias repetitivas repetidas en tándem largas (LTRR), repeticiones cortas espaciadas de manera regular (SRSR), grupos grandes de repeticiones en tándem (LCTR) y repeticiones directas intercaladas con espaciador (SPIDR). En 2002, Jansen y colaboradores las nombraron "CRISPR" y se describieron los genes Cas (Figura 1) (Mojica, Díez-Villaseñor, García-Martínez & Soria, 2005; Barrangou & Horvath, 2017; Jansen, Van Embden, Gaastra & Schouls, 2002). En los siguientes años, se logra entender la función de las múltiples proteínas Cas descubiertas, proponiendo a CRISPR como un sistema de inmunidad adquirida ante los bacteriófagos (Makarova, Grishin, Shabalina, Wolf & Koonin, 2006; Barrangou et al., 2007). Jinek y colaboradores (2012) generan una quimera de RNA que puede unirse a Cas9, sugiriendo una posible aplicación como sistema de edición genética (Jinek et al., 2012). Posteriormente, inicia la revolución CRISPR y su uso se hizo común en bacterias, levaduras, plantas, animales y otros organismos, debido a la rapidez y versatilidad del sistema. En 2020, Jennifer Doudna y Emmanuelle Charpentier de la Universidad de California, Berkeley, recibieron el Premio Nobel de Química por crear y proponer el RNA guía, que marcó el comienzo de la edición genética en todo tipo de células (Ledford & Callaway, 2020). En los siguientes dos años, CRISPR se utilizó para detectar al virus de SARS-CoV-2 mediante pruebas rápidas y lograrlo abatir (de Puig et al., 2021; Liu et al., 2021). Las pruebas portátiles para diagnosticar COVID-19 basadas en CRISPR detectan una amplia gama de objetivos moleculares de importancia médica (Kaminski, Abudayyeh, Gootenberg, Zhang & Collins, 2021). CRISPR también tuvo éxito en intervenciones médicas al insertarlo de manera directa en la retina del ojo humano con el fin de combatir el trastorno de ceguera hereditario (Ledford, 2020). Asimismo, se han obtenido resultados alentadores con CRISPR en la primera terapia celular en humanos para intentar erradicar el cáncer pulmonar (He, 2020), incluyendo estudios exitosos in vitro e in vivo en animales modelo para tratar diferentes tipos de cáncer (Katti, Diaz, Caragine, Sanjana & Dow, 2022). En 2021, se comenzaron a realizar ensayos clínicos para usar CRISPR en el tratamiento de pacientes con la enfermedad de células falciformes (Eisenstein, 2021; Kaiser, 2021). En los últimos años, se han desarrollado más herramientas basadas en CRISPR con resultados positivos en el tratamiento de enfermedades a nivel mundial, como el cáncer de mieloma refractario avanzado, el sarcoma metastásico (Stadtmauer et al., 2020), y la fibrosis quística (Geurts et al., 2020). Además, la tecnología CRISPR/Cas tiene un papel importante en campos como la agricultura (Zaidi, Mahas, Vanderschuren & Magdy, 2020), la bioingeniería y la biotecnología (Lau, 2018). En el futuro nuevas formas inimaginables de editar genomas se sumarán a la lista creciente del sistema CRISPR/Cas.
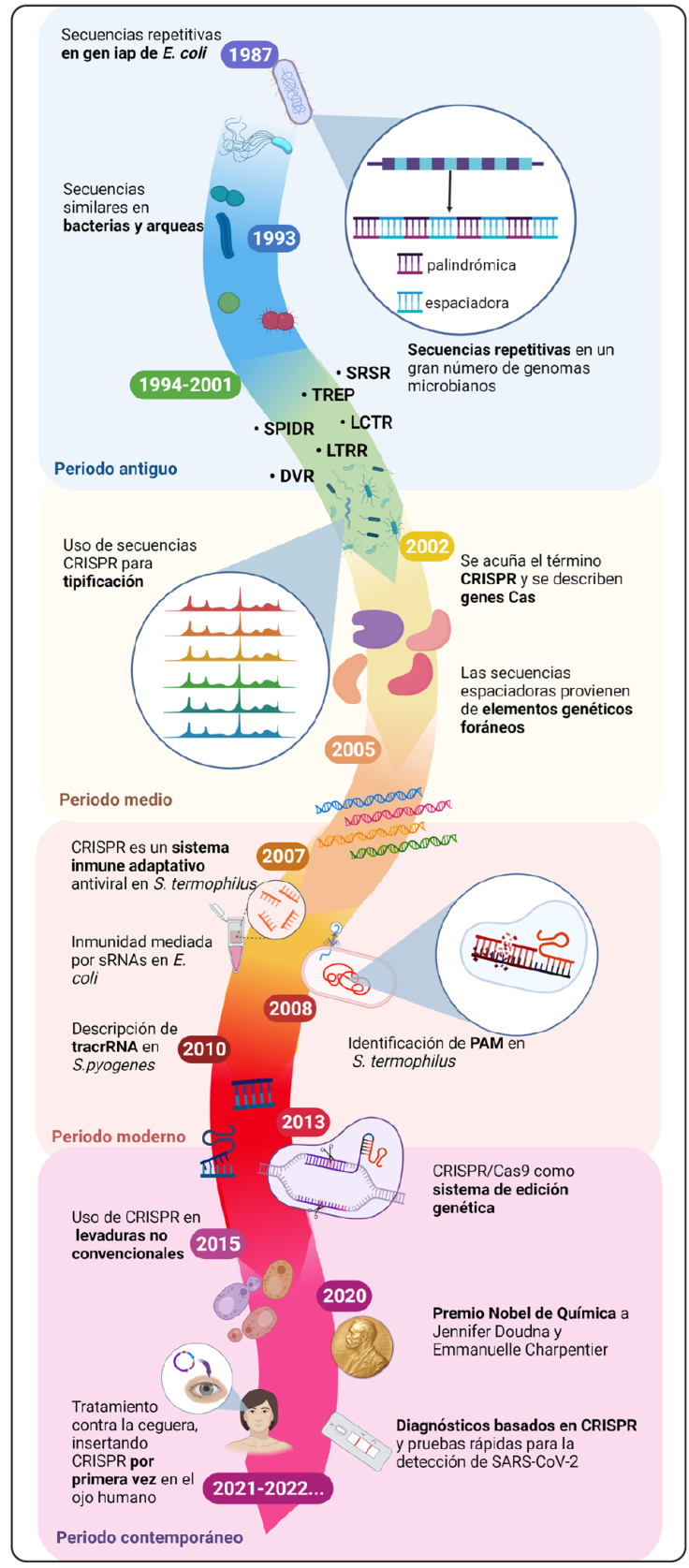
Elaboración personal usando BioRender.com.
Figura 1 Descubrimientos importantes en las investigaciones del sistema CRISPR/Cas. Los periodos antiguo y medio marcan el descubrimiento y elucidación de las misteriosas secuencias CRISPR (1987-2005). Los periodos (moderno y contemporáneo) ilustran los eventos que desarrollaron la famosa tecnología de edición genética CRISPR y sus aplicaciones más novedosas (2007-2022).
El funcionamiento general del sistema CRISPR consta de tres eventos principales: (i) la adquisición de espaciadores, conocida como la fase de adaptación, (ii) la biogénesis del CRISPR RNA (crRNA), y (iii) la interferencia (Figura 2). La adquisición comienza con la detección del DNA foráneo y su integración al cromosoma de la célula en el extremo terminal líder del locus CRISPR. La detección es mediante una secuencia de 2 a 5 nucleótidos llamada motivo adyacente del protoespaciador (PAM o Protospacer Adjacent Motif), identificada en el DNA foráneo con ayuda de las proteínas Cas. Después, la secuencia es añadida en el locus CRISPR como nuevos espaciadores que están flanqueados por secuencias repetidas (Hille et al., 2018). La siguiente etapa consiste en la transcripción del locus CRISPR y los nuevos espaciadores, generando los pre-crRNAs (precursores largos de crRNA), que se convierten en crRNAs maduros con ayuda de las RNAsas y algunas proteínas Cas. Por lo general, el crRNA se mantiene asociado a las proteínas Cas que participaron en su maduración; adicionalmente, en algunos tipos de sistemas CRISPR, el crRNA hibridiza con un RNA no codificante conocido como RNA trans-activado (tracrRNA), que es necesario tanto para el procesamiento del pre-crRNA como para la fase de interferencia (Faure et al., 2019). En la siguiente fase, las nucleasas Cas son guiadas por el crRNA (o bien, el crRNA:tracrRNA) como una “maquinaria de interferencia” hacia el DNA invasor, reconociendo las secuencias PAM, permitiendo el corte en el DNA invasor en las regiones que tienen identidad con la secuencia del crRNA (silenciamiento); o bien, se utilizará este mismo mecanismo para agregar segmentos del DNA invasor en el locus CRISPR y tener un sistema de memoria que ayudará a prevenir futuras infecciones (Hille et al., 2018).

Elaboración personal usando BioRender.com.
Figura 2 Estructura del locus CRISPR/Cas9 y eventos principales del sistema. El locus CRISPR está conformado por los genes que codifican a las proteínas Cas, el tracrRNA y el arreglo de secuencias repetidas (barras grises) intercaladas con secuencias espaciadoras (barras de colores). La secuencia líder se ubica entre el grupo de genes asociados a CRISPR y el arreglo de repeticiones interespaciadas. 1) La adaptación inicia cuando un bacteriófago (rojo) infecta a una célula hospedera por primera vez. Las proteínas Cas (azul) cortan la secuencia del DNA invasor, que será integrada al inicio del arreglo CRISPR del hospedero. 2) La biogénesis del crRNA comienza cuando el arreglo CRISPR es transcrito en pre-crRNA largos (pre CRISPR RNA). Asimismo, los transcritos de los genes tracrRNAs se unen a las secuencias espaciadoras del pre-crRNA; que, en conjunto con las proteínas Cas9 y la actividad de RNAsas, son necesarios para la formación del crRNA maduro. Los crRNAs maduros se mantienen asociados con las proteínas Cas9 para formar los complejos efectores. 3) La interferencia ocurre cuando los bacteriófagos infectan a la célula hospedera por segunda vez, lo que activa a los complejos efectores que reconocen a las secuencias invasoras, guiando a Cas9 a cortar y silenciar el material genético de los invasores.
Los sistemas CRISPR/cas como herramientas moleculares
Una pregunta interesante que se hicieron durante la descripción de este mecanismo fue ¿cómo se adquiere y evoluciona el sistema CRISPR? La respuesta tuvo lugar cuando las investigaciones identificaron numerosos componentes del arreglo CRISPR en distintos microorganismos, que presentaban cambios en cada locus entre grupos y especies. Actualmente, los sistemas CRISPR se dividen en dos grandes clases que contienen 6 tipos: clase 1 (tipos I, III y IV) y clase 2 (tipos II, V y VI) (Koonin & Makarova, 2019). Todos los tipos tienen similitudes y diferencias en las proteínas, los crRNAs, o en los mecanismos de adquisición e interferencia (Figura 2 y Tabla I). El sistema tipo I es el más distribuido en las bacterias y se caracteriza por formar un complejo multiproteico unido al crRNA conocido como “complejo asociado a CRISPR de defensa antiviral” o “Cascade”. Este complejo reconoce las secuencias de DNA invasoras y recluta a una nucleasa Cas3 para hacer el corte correspondiente. El sistema tipo III utiliza los complejos multi-Cas -proteína para la interferencia (Csm). En el sistema tipo II, la endonucleasa Cas9 requiere al tracrRNA para complementarse con el crRNA y, al madurar, reconoce la secuencia PAM de tres nucleótidos del DNA blanco (Shmakov et al., 2017). En el sistema V, la proteína efectora característica es Cas12. Al igual que en los sistemas de tipo II, algunos subtipos de sistemas V requieren el tracrRNA para la maduración del crRNA y la interferencia (Makarova et al., 2020). Por último, el sistema tipo VI utiliza la proteína Cas13, una RNAsa que se activa al unirse al RNA blanco y que degrada tanto al RNA blanco como al RNA colateral. A diferencia de otras proteínas Cas, que reconocen un sitio PAM, Cas13 reconoce un sitio de flanqueo del protoespaciador (PFS) en el RNA invasor (Hille et al., 2018; Koonin & Makarova, 2019).
Tabla I Diferencias entre los sistemas CRISPR clase 1 y 2.
| Tipos | Módulo efector | Endonucleasa | Blanco | Requiere tracrRNA | |
|---|---|---|---|---|---|
| Clase 1 | I | Multiproteico | Cas3 | DNA | No |
| III | Cas10 | DNA/RNA | No | ||
| IV | ? | ? | ? | ||
| Clase 2 | II | Proteína con múltiples dominios | Cas9 | DNA | Sí |
| V | Cas12 | DNA | Algunos subtipos | ||
| VI | Cas13 | RNA | No |
El sistema tipo II es el más utilizado como herramienta molecular para editar genomas de organismos. Este sistema contiene a la endonucleasa Cas9, que genera cortes de doble cadena en el DNA de manera específica guiada por el crRNA, atributo que facilita su uso en la biotecnología (Shmakov et al., 2017).
El crRNA guía (gRNA) madura al formar un dúplex con la secuencia de tracrRNA, codificada río arriba del locus CRISPR/ Cas. El dúplex crRNA:tracrRNA se puede utilizar como una molécula quimérica denominada sgRNA (single guide RNA). El sgRNA dirige de manera específica a Cas9 debido a que contiene tanto a los 20 nucleótidos del cRNA complementarios a la secuencia blanco en el extremo 5’, como a la estructura de doble cadena que se forma en el extremo 3' del sgRNA. Una vez maduro, el dúplex crRNA:tracrRNA es requerido para que Cas9 se dirija hasta el sitio complementario del DNA invasor, llevando a cabo la fase de interferencia; se ha demostrado que el tracrRNA es indispensable para que funcione el sistema, ya que en su ausencia se pierde la actividad de Cas9 (Doudna & Charpentier, 2014; Jinek et al., 2012).
En general, los sistemas CRISPR requieren del reconocimiento de una secuencia PAM, tanto para identificar a los protoespaciadores en la fase de adquisición como para dirigir a la maquinaria de interferencia. La secuencia específica del motif varía de acuerdo con la especie del hospedero del sistema CRISPR/ Cas. La secuencia PAM que reconoce Cas9 de la bacteria Streptococcus pyogenes es NGG y para realizar cortes de doble cadena específicos de manera dirigida sólo es necesario cambiar la secuencia de 20 nt del espaciador en el crRNA a una secuencia adyacente a un sitio PAM. Los dominios tipo HNH y RuvC de Cas9 llevan a cabo el corte en cada una de las cadenas del DNA. Cuando se realizan mutaciones en alguno de los dos dominios, Cas9 realiza cortes en una sola cadena del DNA; a esta variante se le conoce como Cas9 nickasa (nCas9), mientras que las mutaciones en ambos dominios provocan la pérdida de actividad de la nucleasa de Cas9; sin embargo, se sigue uniendo al DNA de manera específica y se conoce como dead Cas9 (dCas9) (Doudna & Charpentier, 2014; Jinek et al., 2012). En su ambiente nativo, Cas9 lleva a cabo la fase de interferencia en la respuesta inmune de S. pyogenes. Durante la invasión por bacteriófagos, se expresa el cassette CRISPR produciendo múltiples crRNAs que guían a Cas9 hacia el DNA invasor y lo cortan (Figura 2). Con la programación de Cas9 fue posible usar el sistema como herramienta molecular en la edición genética de manera precisa y dirigida en organismos que incluso no son modelo y aprovechar los mecanismos de reparación del DNA nativo.
Edición genética de levaduras en la era de CRISPR
Las levaduras son hongos unicelulares con aplicaciones biotecnológicas en las áreas de medio ambiente (biorremediación y degradación de contaminantes), biocontrol (protección de cultivos, seguridad agrícola y probióticos), investigación en ciencias biomédicas (descubrimiento de fármacos, metabolismo, resistencia a fármacos y elucidación de mecanismos de las enfermedades), investigación básica en ciencias biológicas (biología celular y molecular, genómica comparada y funcional, ingeniería de rutas metabólicas y biología de sistemas), producción de proteínas (proteínas para uso farmacéutico, enzimas, hormonas, vacunas y toxinas), biocatálisis (productos farmacéuticos, productos químicos y biotransformadores), alimentos e ingredientes (enzimas, saborizantes, pigmentos, aminoácidos y ácidos orgánicos) y energías renovables (producción de biocombustibles y lipasas) (González et al., 2020; Segal-Kischinevzky et al., 2022). Durante la bioproducción con frecuencia se recurre a la ingeniería genética de levaduras que incluye la eliminación de genes, la sobreexpresión de proteínas o expresión heteróloga, lo que permite la optimización de la síntesis de metabolitos con valor agregado. Por lo tanto, el desarrollo de métodos y herramientas moleculares a partir de la edición genética de las levaduras con potencial biotecnológico es de gran interés para la academia y la industria.
Las primeras investigaciones que usaron ingeniería genética en la levadura Saccharomyces cerevisiae se remontan a principios de 1980, aunque su genoma se terminó de secuenciar en 1996 (Fraczek, Naseeb & Delneri, 2018). La mayoría de las herramientas moleculares de edición genética se han desarrollado en S. cerevisiae por ser de fácil manipulación, su trazabilidad genética y su genoma bien anotado (Wagner & Alper, 2016). Históricamente, la manipulación genética en S. cerevisiae es eficiente debido a su alta tasa de reparación dirigida por identidad (HDR, Homology Directed Repair). La edición genética en S. cerevisiae se realiza a través de la construcción de módulos de DNA generados por PCR y la integración de estos en su genoma mediante diferentes métodos de transformación (Wach, Brachat, Pöhlmann & Philippsen, 1994). Otros métodos de manipulación genética en levaduras que se basan también en la recombinación homóloga son: (i) el sistema de recombinación Cre-Lox, que hace uso de la recombinasa sitio-específica Cre y el reconocimiento de secuencias Lox que flanquean al gen de interés, (ii) el sistema Delitto perfetto, que utiliza de múltiples oligonucleótidos para modificar al gen de interés, y (iii) el empleo de meganucleasas, que son enzimas de restricción con alta especificidad al sitio que reconocen. Sin embargo, estos sistemas tienen desventajas porque la mutagénesis está restringida a la región del cassette integrado (Fraczek et al., 2018).
Las nuevas tecnologías de manipulación genética como CRISPR/Cas tienen la capacidad de superar estos obstáculos y generar nuevas herramientas moleculares eficientes, que han revolucionado a la ingeniería genética de levaduras en las últimas décadas (Shan, Dai & Wang, 2021). La manipulación genética mediante CRISPR/Cas9 se ha hecho de manera exitosa tanto en S. cerevisiae como en otras levaduras del género Candida (incluyendo a C. albicans), Cryptococcus neoformans, C. deneoformans, Yarrowia lipolytica, Pichia pastoris, entre otras. El sistema CRISPR/Cas9 se ha utilizado de tres formas: i) inserción de uno o más vectores de expresión con el cassette de Cas9 y una o más secuencias de gRNA, integrando al genoma de la levadura el cassette de Cas9, ii) expresión del gRNA desde un plásmido, y iii) inserción de la proteína Cas9 en complejo directo con las moléculas de gRNA de manera transitoria, sin requerir de la expresión del sistema (Arras et al., 2016; Grahl, Demers, Crocker & Hogan, 2017; Fraczek et al., 2018). En las primeras dos estrategias se expresa de manera constitutiva a Cas9, utilizando el promotor del gen de elongación de la traducción TEF1. Por otro lado, la sobreexpresión de Cas9 con el promotor de los genes que codifican para el transportador de la glucosa de alta afinidad y la gliceraldehído 3-fosfato deshidrogenasa (HXT7 o TDH3), resultó en efectos negativos para el crecimiento del cultivo y/o en la eficiencia de la transformación. Asimismo, las construcciones con promotores débiles resultaron en bajas eficiencias de edición genética (Raschmanová, Weninger, Glieder, Kovar & Vogl, 2018). Por lo tanto, ahora se sabe que es necesario elegir promotores específicos que expresen de forma constitutiva a Cas9 sin efectos negativos.
La implementación del sistema CRISPR/Cas9 se ha estandarizado mediante la transformación con un vector, que contiene tanto la secuencia que codifica para Cas9 como la secuencia del gRNA. Asimismo, se requiere la secuencia del molde o módulo de reparación, que puede estar codificada en el mismo vector, en otros vectores individuales, o se puede generar de manera independiente mediante PCR.
La modificación genética en levaduras que pertenecen al clado CTG se ha logrado al utilizar moldes de reparación que contienen la secuencia del gen NAT1 o SAT1 (confieren resistencia al antibiótico nourseotricina o estreptomicina, respectivamente), que está optimizada para evitar el uso del codón CTG. Esta estrategia se ha utilizado para mutar al gen ADE2, involucrado en la síntesis de la adenina (Min, Ichikawa, Woolford & Mitchell, 2016; Norton, Sherwood & Bennett, 2017; Vyas, Barrasa & Fink, 2015).
Grahl y colaboradores (2017) desarrollaron un sistema de edición genética con CRISPR/Cas9 libre de expresión para C. lusitaniae, C. glabrata y C. auris, el cual funciona en otros organismos (hongos filamentosos, plantas, algas, células de mamíferos, ratones y líneas celulares humanas). Este sistema consiste en formar un complejo in vitro (gRNA/Cas9) que después se introduce en la levadura mediante electroporación, para generar cortes de doble cadena de DNA en la región blanco. La electroporación se puede realizar en presencia de un cassette o módulo de recombinación que contiene al gen NAT1, el cual se inserta en la región blanco (Grahl et al., 2017). Basado en este sistema, Wang, P., usó la proteína Cas9 en complejo con los gRNA para realizar un knockout del gen GIB2 (proteína tipo Gβ/RACK1) en la levadura Cryptococcus neoformans (Wang, 2018).
A partir de 2012, el sistema de tipo II característico por usar Cas9 recibió un enorme interés debido a la revelación del funcionamiento de esta nucleasa y su alta tasa de eficiencia, lo que de inmediato sugirió posibles aplicaciones en la ingeniería genética. A inicios de 2013 se logró demostrar que este sistema realiza mutaciones específicas en el DNA de células procariotas y eucariotas, tan solo con expresar de manera heteróloga la proteína Cas9 junto con un RNA guía (sgRNA) complementario a la región que se desea editar en el genoma. El sgRNA funciona como crRNA maduro (es decir un complejo crRNA:tracRNA) con una parte estructural y otra variable de 20 pares de bases (pb) que coincide con la región que se desea editar (Sternberg & Doudna, 2015; Raschmanová et al., 2018). Posteriormente, este sistema se implementó de manera exitosa en la levadura modelo S. cerevisiae (DiCarlo et al., 2013), que antes ya había sido editada genéticamente con la recombinación mediada por Cre-Loxp (Sauer, 1987), el método de “Delitto Perfetto” (Storici, Lewis & Resnick, 2001) y el uso de meganucleasas, que son técnicas costosas y requieren el doble de tiempo que la edición mediante CRISPR (Tabla II).
Tabla II Ventajas y desventajas de las estrategias de edición genética en levaduras.
| Método de edición genética | Ventajas | Desventajas | Optimización |
|---|---|---|---|
| Cre-LoxP | Eficiente en levaduras que utilizan la HR con mayor frecuencia (e.g., S. cerevisiae). | Poco versátil, consume demasiado tiempo y es costoso. | - |
| Delitto Perfetto | Eficiente y preciso en levaduras que utilizan la HR con mayor frecuencia (e. g., S. cerevisiae). | Poco versátil y usa múltiples oligonucleótidos. | - |
| Meganucleasas | Preciso cuando hay un sitio de corte específico. | Poco versátil, usa muchas nucleasas que encarecen los ensayos. | - |
| CRISPR/Cas | Versátil en múltiples especies, altamente eficiente, usa pocos componentes. | La sobreexpresión de Cas9 puede generar mutaciones no deseadas (off target). |
|
Existen algunos aspectos fundamentales que se deben considerar para la edición y reparación del corte de doble cadena del DNA mediante el sistema CRISPR/Cas9 (Figura 3). La reparación del corte es realizada de dos formas: la vía de recombinación homóloga (HR) que requiere un DNA molde con identidad a la secuencia dañada para corregir de manera correcta mediante su integración, o bien, por la unión de extremos no homólogos (NHEJ), que no requiere de un molde porque la reparación del corte se logra de manera inmediata por ligación. No obstante, durante el proceso de reparación se pueden generar inserciones y/o deleciones de nucleótidos (indeles, inserciones-deleciones) (Fraczek et al., 2018; Shan et al., 2021). Se ha observado que la HR resulta muy eficiente en S. cerevisiae; sin embargo, en otras levaduras predomina la NHEJ sobre la HR, complicando la edición genética y como resultado se obtienen fenotipos no deseados (Raschmanová et al., 2018). Por lo tanto, la implementación del sistema CRISPR/Cas9 en levaduras no convencionales resulta desafiante, pero prometedora.

Elaboración personal usando BioRender.com.
Figura 3 Tecnología de edición genética CRISPR/Cas9 en levaduras. A) Se transforma a la levadura con un vector que expresa los componentes del sistema CRISPR (Cas9 y sgRNA). La nucleasa Cas9 (azul) y el sgRNA (naranja-gris) forman un complejo que se une a la secuencia blanco del DNA, cortando la doble cadena adyacente al sitio PAM (verde). B) La reparación puede ocurrir por la vía de unión de los extremos no homólogos (NHEJ) que liga extremos y genera inserciones y/o deleciones (rojo). El corte también puede ser reparado por la vía de recombinación homóloga (HR), que requiere un molde de DNA con la modificación o mutaciones (rosa) que se desea integrar.
Implementación de las tecnologías CRISPR en las levaduras
Existe una vasta documentación sobre la implementación del sistema CRISPR/Cas en S. cerevisiae. Esta herramienta molecular ha permitido incorporar y/o remover secuencias, activar o reprimir genes, etc. (Shan et al., 2021). A continuación, se describen algunas investigaciones destacadas que lograron adaptar de manera exitosa el sistema CRISPR en distintas levaduras con potencial biotecnológico.
Generación de un knockout
La eliminación de la función de un gen o una región genómica se logra al generar mutaciones o removiendo secuencias de interés, utilizando a Cas9 o Cas12 y un sgRNA que guíe a la endonucleasa hacia un sitio específico de corte de doble cadena en la secuencia blanco del DNA. La reparación del corte dependerá de la capacidad del organismo; sin embargo, durante la misma se pueden introducir mutaciones o deleciones, con pérdida de la función y generar un knockout (Ko). Este tipo de mutaciones permite identificar la función de las regiones codificantes y regiones no codificantes con alguna función.
La mayor dificultad para generar un Ko en levaduras no convencionales es la falta de métodos y protocolos de transformación eficientes. Por ejemplo, en la levadura halotolerante Debaryomyces hansenii predomina la NHEJ como vía de reparación del DNA, que dificulta mutar sus genes. Sin embargo, Spasskaya y colaboradores (2021) lograron eliminar el gen que codifica para la fosforribosil aminoimidazol carboxilasa de la ruta de biosíntesis de la adenina (ADE2) mediante CRISPRKo. Utilizando la endonucleasa Cas9 y un sgRNA (Figura 4), obtuvieron la disrupción de este gen en hasta un 80% de las colonias transformadas. De igual manera, probaron agregar un templado de DNA para favorecer la reparación por HR, aumentando la eficiencia de eliminación del gen hasta un 90% con un sgRNA. Para remover el gen completo utilizaron dos sgRNAs, y se observó que el 38% de las colonias transformadas fueron Ko. Generar un Ko del gen ADE2 tiene una gran ventaja para seleccionar las mutantes, ya que la eliminación de este gen puede ser fácilmente observada en el fenotipo de las colonias transformadas (WT, colonias blancas / mutante ade2, colonias rojas) (Spasskaya et al., 2021). Tiempo después, Strucko y colaboradores (2021) interrumpieron la NHEJ con la finalidad de generar mutaciones puntuales o eliminar genes a través de la HR sin el uso de marcadores (Strucko et al., 2021).

Elaboración personal usando BioRender.com.
Figura 4 Tecnología de CRISPR/Cas9 knockout en la levadura D. hansenii. 1) Se transforma a la levadura con un plásmido que contiene al gen que codifica para Cas9 y un sgRNA que guíe a la endonucleasa hacia una región del gen ADE2. Cas9 (azul) realiza un corte de doble cadena que puede generar mutaciones (indeles representados con una estrella) durante la reparación NHEJ. La inactivación del gen ADE2 provoca que las células adquieran una coloración roja, que facilita distinguir y seleccionar a las colonias que contienen la mutación. 2) Se transforma a la levadura con un plásmido que contenga al gen de Cas9 y dos sgRNA, los cuales dirigen a Cas9 hacia las regiones intergénicas 5’ y 3’ del gen ADE2 provocando su completa eliminación (tachado en rojo). Las flechas horizontales simbolizan la transcripción del gen. Las flechas de colores indican los sitios de corte.
Estrategias de edición multiplex
En los últimos años, la utilización de los sistemas de edición genética CRISPR se ha incrementado con el fin de realizar mutaciones o deleciones en genes o en regiones puntuales del genoma (McCarty, Graham, Studená & Ledesma-Amaro, 2020). No obstante, este enfoque individual supone limitaciones importantes en cuanto a la eficiencia y a las aplicaciones biotecnológicas de los sistemas CRISPR, pues muchas veces es necesario editar múltiples regiones genómicas para modificar e introducir varios genes con el fin de duplicar algunas vías metabólicas de interés (Adiego-Pérez et al., 2019). Repetir el proceso de edición individual varias veces, no parece ser lo más adecuado; por fortuna, el sistema de edición genética CRISPR/Cas9 posee la ventaja de editar múltiples regiones del genoma durante una sola transformación, a diferencia de otros sistemas de edición genética, con la implementación de varios gRNA distintos junto con la proteína Cas9 (Minkenberg, Wheatley & Yang, 2017).
La tecnología de edición multiplex basada en el sistema CRISPR, es una herramienta versátil que puede ser empleada para modificar dos o más regiones del genoma de manera simultánea y precisa, posibilitando la eliminación, activación o represión de genes, e incluso la modificación del epigenoma (Abdelrahman, Wei, Rohila & Zhao, 2021; Minkenberg et al., 2017). Existen diferentes estrategias generales utilizadas para la expresión de un sgRNA multiplex (Utomo, Hodgins & Ro, 2021). La estrategia más conocida y utilizada es la denominada cassette de expresión de un sgRNA de promotor individual, que emplea un cassette que contiene dos o más gRNA que incluyen un promotor y un terminador (Cao, Xiao & Yan, 2018). Un ejemplo de esta estrategia puede revisarse en el trabajo de Otoupal y colaboradores (2019), quienes investigaban cómo potenciar las capacidades de producción de los bioproductos de la levadura Rhodosporidium toruloides mediante la implementación de CRISPR/Cas9 como herramienta de edición genética. Para ello, diseñaron cuatro sgRNAs en dos construcciones individuales de Cas9/sgRNA dirigidas a: (i) el gen que codifica para la fitoeno sintasa/licopeno ciclasa (CAR2, esencial en la biosíntesis de carotenoides), y (ii) el gen que codifica para la enzima involucrada en la síntesis de novo del ribonucleótido de pirimidina (URA3, esencial en la biosíntesis de uracilo). Ambas construcciones se implementaron de modo que la escisión se produjera en dos sitios separados de ~500 pb en ambos genes de la levadura R. toruloides (Figura 5). Los fenotipos obtenidos tras la transformación fueron los cambios en el color de la levadura de un tono rojizo a un tono blanquecino, evidenciando la pérdida de la capacidad de síntesis de los carotenoides. El análisis de los resultados indicó que la interrupción del gen CAR2 en una sola transformación podía alcanzar hasta un 50% de la eficiencia de edición, mientras que para el gen URA3, la eficiencia de edición después de ser optimizada estuvo por arriba del 0.6%, demostrando que la edición multiplex funciona para editar el genoma de R. toruloides (Otoupal et al., 2019). Otra estrategia utilizada para la edición multiplex consiste en emplear sgRNAs que se codifican y procesan mediante mecanismos basados en los sistemas nativos de CRISPR/Cas. En este sistema se introducen Cas9, tracrRNA y pre-crRNA; este último es transcrito a partir de un arreglo de crRNAs en un solo vector de expresión. Al igual que en el sistema nativo de CRISPR/Cas, los crRNAs pueden ser procesados por una RNAsa III dependiente de tracrRNA. Sin embargo, al emplear tracrRNA para imitar el sistema nativo de CRISPR, disminuye la eficiencia, razón por la que esta estrategia no es muy recomendable (McCarty et al., 2020). Un tercer método que es más efectivo consiste en la implementación de Cas12a (Cpf1) o Cas13a, que carece del requisito de emplear tracrRNAs para madurar los pre-crRNAS. Este método aprovecha la capacidad intrínseca de las Cas para escindir el pre-crRNA a través del reconocimiento de sus estructuras secundarias de horquilla, permitiendo la formación de crRNAs maduros y así procesar un gran número de sgRNAs (Zetsche et al., 2017; McCarty et al., 2020).
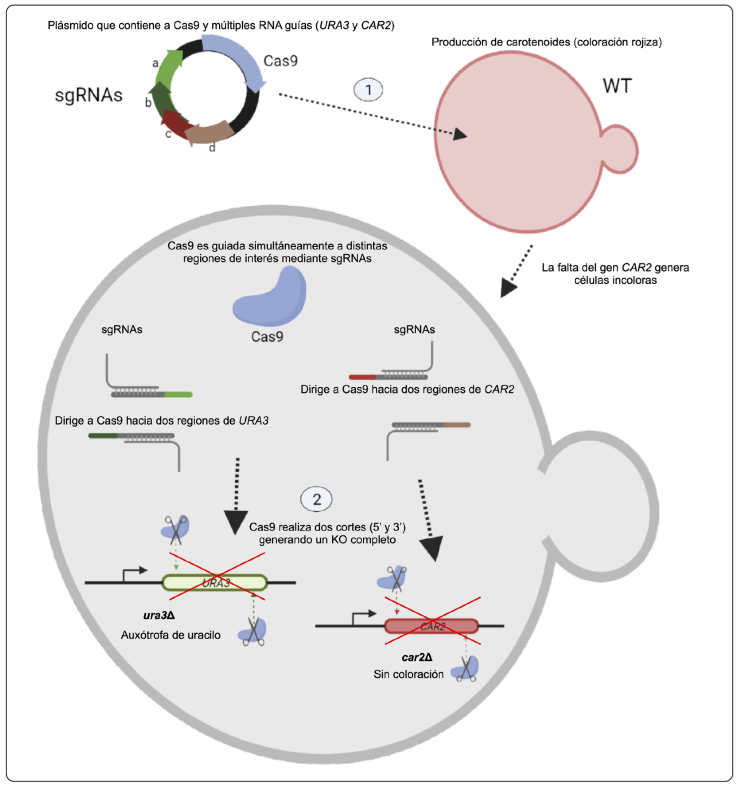
Elaboración personal usando BioRender.com.
Figura 5 Tecnología de integración multiplex CRISPR/Cas9 en la levadura R. toruloides. 1) Se transforma la cepa WT (una levadura pigmentada por la producción de carotenoides que tiene un fenotipo rojizo) con un plásmido que contiene los cassettes que expresan múltiples sgRNAs complementarios a los genes URA3 y CAR2. Los módulos “a” y “b” transcriben los sgRNAs complementarios al gen URA3, mientras que los módulos “c” y “d” transcriben los sgRNAs complementarios al gen CAR2. 2) Los sgRNAs “a” y “b” guían a Cas9 (azul) hacia las dos regiones del gen URA3 que serán escindidas (verde). Del mismo modo, la expresión de los sgRNAs “c” y “d” permiten la escisión de dos regiones del gen CAR2 (marrón). Después de la integración multiplex, como marcador de selección, el fenotipo de la levadura cambia de una coloración rojiza a una coloración blanquecina o sin color. Las flechas horizontales negras simbolizan la transcripción de los genes. Las flechas de colores indican los sitios de corte.
Edición de genomas por medio de integración multiplex
La mayoría de los trabajos que aplican la tecnología multiplex se basan en la preparación de vectores de expresión que buscan generar mutaciones puntuales o inactivar múltiples genes en específico. No obstante, para algunos fines resulta conveniente buscar la integración del constructo multiplex en el genoma del organismo con el que se desea trabajar (Horwitz et al., 2015). Aunque existen diferentes estrategias metodológicas para realizar una integración genómica como las nucleasas efectoras de tipo activador de la transcripción (TALEN), la edición multiplex asistida por TALEN (TAME) o las nucleasas con dedos de zinc (ZFN), los científicos han optado por utilizar el sistema de edición basado en CRISPR/Cas, debido a que proporciona un enfoque más simple, barato, versátil y con mayores tasas de éxito (Montecillo, Chu & Bae, 2020).
La edición del genoma por multiplex es una técnica que permite reunir múltiples genes de interés en una sola transformación dentro del genoma de una célula (Utomo et al., 2021). Malcı y colaboradores (2020) realizaron una revisión sobre los principales métodos de integración multiplex al genoma basado en CRISPR/Cas en levaduras. En su investigación destacan los métodos de Genome-Specific Multiplex Integration, Donor DNA Delivery Options, y Pre-Placed Gate Systems (Malcı, Walls & Ríos-Solís, 2020). El método de Genome-Specific Multiplex Integration primer se basa en la integración del sistema CRISPR/Cas9 conocido como método de integración delta (δ), e implica el uso de un retrotransposón de levadura (Ty), que tiene dos repeticiones directas terminales o secuencias δ. Las secuencias génicas heterólogas se insertan en la secuencia δ. La integración de CRISPR/Cas9 permite inducir múltiples rupturas de doble cadena en las secuencias δ, al admitir la integración simultánea de varias copias de grandes secuencias de DNA mediante HR (Malcı et al., 2020). El método Pre-Placed Gate Systems consiste en el uso de fragmentos sintéticos introducidos previamente y que sirven como puntos de inserción para la integración de genes. Los sgRNA deben reconocer estos sitios para facilitar la recombinación homóloga (Hou, Qin & Dai, 2018). Por último, el método Donor DNA Delivery Options se basa en la integración de plásmidos, con la ventaja de realizar cortes de doble cadena mediante CRISPR/Cas9 para mejorar la eficiencia de la integración multiplex en el genoma a través de la HR (Liu et al., 2019). El método consiste en integrar a Cas9 en el genoma mientras que los sgRNAs se expresan a partir de un vector integrador linealizado (Malcı et al., 2020). Un ejemplo de este último método fue implementado por Horwitz y colaboradores (2015), en la levadura Kluyveromyces lactis, con la finalidad de probar la eficacia del método montado previamente en S. cerevisiae. Para este experimento, integraron una vía condensada del ácido mucónico, que está constituida por seis genes (AroY, AroZ, AroD, CATA, AroF y AroB), divididos en tres construcciones de integración que son guiadas a los loci DIT1, ADH1 y NDT80 (Figura 6). Adicionalmente, la cepa K. lactis ATCC 8585 fue transformada con Cas9 en el locus GAL80. Al finalizar los experimentos de transformación, los autores consiguieron la integración sin marcadores de las tres construcciones en el genoma de K. lactis en un solo paso, con una triple integración eficaz. Este método permite realizar pruebas rápidas de combinación de mutaciones, así como la creación de prototipos de rutas metabólicas completas, que aceleran de manera drástica la ingeniería de cepas para la investigación básica y aplicada en la fermentación industrial (Horwitz et al., 2015).
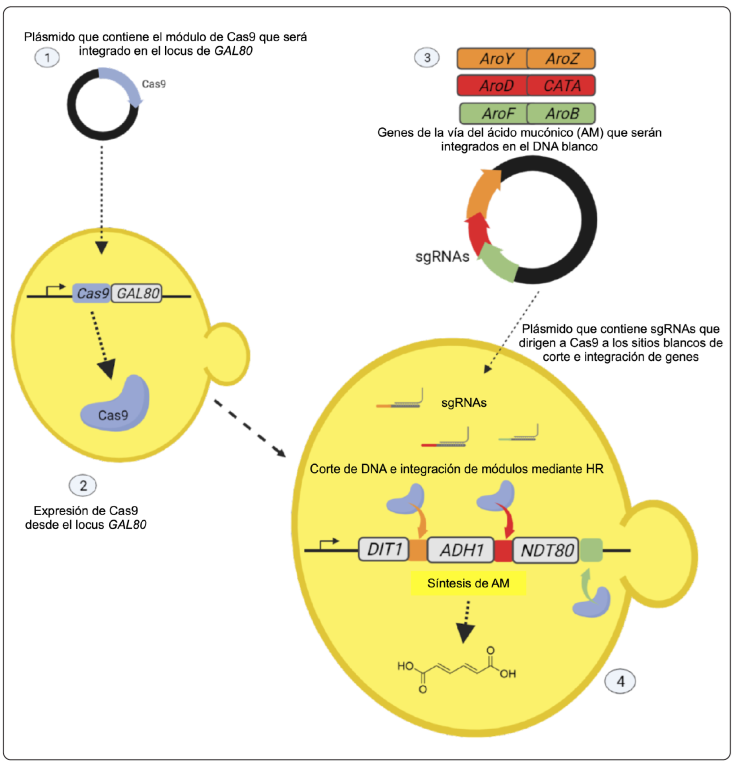
Elaboración personal usando BioRender.com.
Figura 6 Tecnología multiplex en el genoma basado en CRISPR/Cas9 usando como ejemplo la integración de la vía del ácido mucónico en la levadura K. lactis. 1-2) Se transforma a la levadura con un plásmido que contiene el cassette para integrar a Cas9 en el locus de GAL80. 3) Se transforma a la levadura con un segundo plásmido que contiene un cassette con los genes AroY, AroZ, AroD, CATA, AroF y AroB (vía del ácido mucónico, AM), que están distribuidos en tres construcciones (naranja, roja y verde) y expresan tres sgRNAs que se integran en los loci DIT1, ADH1 y NDT80 tras la inducción de los cortes de doble cadena con Cas9 (azul) y la posterior reparación por HR. Las flechas horizontales negras simbolizan la transcripción de los genes. Las flechas de colores indican cada sitio de corte y la región de integración de cada módulo.
Regulación de la transcripción por CRISPR de interferencia o activación
Los reguladores globales de la transcripción controlan múltiples genes en un organismo y hasta la fecha es difícil utilizarlos para regular la transcripción de un solo gen de forma específica. Actualmente, es posible modular la expresión de un gen mediante la utilización de una Cas9 inactiva y un sgRNA apropiado que en conjunto interfieren en los procesos de elongación transcripcional, la unión de las RNA polimerasas o factores de transcripción de manera específica. Esta incapacidad de Cas9 de funcionar como endonucleasa (dCas9 o Dead Cas9) ocurre debido a dos mutaciones puntuales (D10A y H840A) en los dominios RuvC y HNH, que son los responsables del corte en la doble cadena del DNA, lo cual evita que Cas9 realice cortes en el DNA, sin perder la capacidad de ser dirigida a un sitio específico en el genoma (Löbs, Schwartz & Wheeldon, 2017; Qi et al., 2013; Zheng, Su & Qi, 2019). Se ha observado que dirigir dCas9 hacia el promotor de un gen puede bloquear físicamente a la maquinaria de la RNA polimerasa, interfiriendo y suprimiendo la transcripción. Esta herramienta se conoce como CRISPR interference (CRISPRi). Por el contrario, la herramienta CRISPR activation (CRISPRa) utiliza un activador transcripcional fusionado a dCas9 que aumenta la expresión del gen blanco (Zheng et al., 2019). Román y colaboradores (2019) utilizaron este sistema para modular la expresión génica de C. albicans mediante la integración de dCas9 en su genoma bajo el control del sistema inducible TET OFF (Román, Coman, Prieto, Alonso-Monge & Pla, 2019). De esta manera, controlaron la expresión del gen que codifica para la catalasa (CAT1) y fusionaron al gen de la proteína verde fluorescente (GFP) con la doxiciclina como señal de inhibición de la expresión de dCas9 y sus sgRNAs, evitando que dCas9 se traduzca y obstruya al promotor del gen (pCAT1 o P CAT1 ). Por lo tanto, el sistema inducible permite modular la expresión de un gen en un momento específico. La represión transcripcional específica por CRIPSRi también se puede lograr por la expresión del dCas9 fusionada al represor Nrg1 (dCas9-Nrg1) y un sgRNA que tiene identidad con pCAT1 (Figura 7).

Elaboración personal usando BioRender.com.
Figura 7 Tecnología de represión transcripcional CRISPR/dCas9-Nrg1 en la levadura C. albicans. Se transforma a la levadura con un plásmido que contiene la secuencia de dCas9 (gris) y un sgRNA (naranja) para dirigirla hacia la región promotora del gen CAT1 (pCAT1) que codifica para una catalasa. El represor Nrg1 (rojo) se encuentra fusionado a dCas9 (dCas9-Nrg1), que reprime la expresión del gen CAT1 fusionado a la secuencia de la proteína verde fluorescente (GFP). La represión disminuye la transcripción de CAT1-GFP, reflejada en la disminución de la intensidad de la fluorescencia. La flecha horizontal negra indica la transcripción del gen.
Por otra parte, la activación transcripcional por CRISPRa se logra con un sistema de doble activación que utiliza dCas9 fusionada al activador transcripcional Gal4 y un sgRNA modificado que recluta a los activadores de la transcripción (Figura 8). La modificación es un módulo agregado al sgRNA en el extremo 3’ de la molécula en forma de una estructura tallo-asa, que es reconocida por una proteína de unión al RNA fusionada a su vez con los factores de transcripción, como VP64 en el módulo MCP-VP64. Esta estrategia permite acoplar la actividad de CRISPRi o CRISPRa a un solo cassette de expresión, y tiene como ventaja modificar sólo la secuencia del sgRNA para activar o reprimir uno o más genes en el organismo (Zalatan et al., 2015).

Elaboración personal usando BioRender.com.
Figura 8 Tecnología de activación transcripcional CRISPR/dCas9 en la levadura C. albicans. Se transforma a la levadura con un plásmido que contiene la secuencia de dCas9-Gal4 y sgRNA-VP64, este complejo es capaz de incrementar la expresión del gen CAT1 (catalasa) que está fusionado al gen de la proteína verde fluorescente (GFP). El complejo dCas9-Gal4-sgRNA es guiado al promotor de CAT1 (pCAT1) y aumenta la transcripción de CAT1-GFP. Asimismo, el sgRNA contiene una modificación que permite reclutar al activador transcripcional VP64 (compuesto por cuatro copias en tándem de la proteína viral del herpes simple 16, VP16) y la proteína de unión (MCP). Por lo tanto, el gen CAT1 incrementa su expresión en presencia de VP64-MCP y Gal4 reflejado en la detección de la intensidad de la fluorescencia de la GFP.
Edición genética a través de CRISPR Nickase
En el sistema CRISPR Nickase se utiliza una variante de Cas9 que genera un corte de hebra única, o nick, en el DNA (nickasa Cas9, nCas9). Este sistema ofrece la posibilidad de editar otras regiones además de las que se encuentran fuera del rango de la secuencia del sgRNA, a una distancia de hasta 59 pb del sitio del corte de la nCas9. Asimismo, a diferencia de los cortes de la doble hebra, los nicks inducen de manera preferencial a la reparación mediante HR sobre la vía de NHEJ, y disminuyen las inserciones y deleciones innecesarias en el sitio del corte de nCas9 (Satomura et al., 2017) (Figura 9).

Elaboración personal usando BioRender.com.
Figura 9 Tecnología de CRISPR Nickase. 1) Se transforma a la levadura con un plásmido que contiene los cassettes de expresión de nCas9 (morado) y el sgRNA. La secuencia del DNA que se usará como molde para la HR puede estar incluida en el cassette. 2) Una nCas9 genera los cortes de hebra única al tener inactivo uno de sus dos dominios (HNH o RuvC) con actividad de endonucleasa. La nCas9 corta a 3 pb río arriba del PAM. 3) La secuencia blanco es reparada vía HR utilizando un molde de DNA que, además de contener las regiones de homología con la secuencia editada, introduce una mutación (en rojo). El sistema CRISPR Nickase permite introducir la mutación, ya sea dentro o fuera de las regiones reconocidas por el sgRNA, así como fuera o dentro del PAM, a una distancia de hasta 59 pb del sitio del corte de la nickasa.
Como se mencionó, la endonucleasa Cas9 posee dos dominios activos con actividad de endonucleasa (HNH y RuvC). El dominio HNH corta a la hebra del DNA que contiene a la secuencia blanco (complementaria a la secuencia de sgRNA), mientras que el dominio RuvC corta a la otra hebra del DNA. Jinek y colaboradores (2012) desarrollaron variantes de Cas9 con mutaciones puntuales que inactivan la actividad catalítica de HNH y RuvC por separado. La variante H840A (His840→Ala840) corresponde a la forma inactiva de HNH, entre tanto D10A (Asp10→Ala10) es la forma inactiva de RuvC. Al inactivar a cualquiera de estos dos dominios y dejar sólo uno activo, se inducen cortes de hebra única, o nicks, en el DNA (Jinek et al., 2012). Por otra parte, Gasiunas y colaboradores (2012) también desarrollaron mutantes de Cas9 con dominios HNH o RuvC inactivos (las mutantes N891A y D31A, respectivamente) y obtuvieron nicks en el DNA, corroborando que se necesita que ambos dominios se encuentren activos para inducir cortes de doble cadena en el DNA (Gasiunas, Barrangou, Horvath & Siksnys, 2012).
En las levaduras, los nicks pueden ser reparados por HR en una vía que depende sobre todo de las DNA polimerasas β, δ ó ε (Caldecott, 2014; Vriend & Krawczyk, 2017), sin necesidad de algunas de las proteínas accesorias implicadas en la reparación de los nicks en las células de vertebrados, como PARP1 y XRCC1 (Semighini, Savoldi, Goldman & Harris, 2006). Sin embargo, no fue sino hasta el desarrollo del sistema CRISPR Nickase que fue posible empezar a investigar cuáles son las enzimas clave en la reparación de los nicks por recombinación homóloga en las levaduras (Satomura et al., 2017; Smith et al., 2009). El trabajo de Satomura y colaboradores (2017) aprovechó el sistema CRISPR Nickase desarrollado por ellos mismos, para incluir un experimento con mutantes en Rad51 y Rad52, unas proteínas que participan en la reparación de los cortes de doble cadena en S. cerevisiae. Estos experimentos permitieron observar que el sistema Nickase sólo podía editar al genoma si Rad52 que se encontraba expresada y fosforilada, por lo que sugirieron que la reparación de los nicks por HR en levaduras es dependiente de Rad52p e independiente de Rad51p (Satomura et al., 2017).
Los grupos de investigación de Jinek (2012) y Gasiunas (2012) evidenciaron que los dominios HNH y RuvC generan nicks en el DNA de manera independiente (Gasiunas et al., 2012; Jinek et al., 2012). El grupo de investigación de Satomura (2017), desarrolló un plásmido que contenía los cassettes de la nCas9, el sgRNA y el DNA donador para introducir un codón de paro en el gen que confiere resistencia a la canavanina (CAN1) en S. cerevisiae (Satomura et al., 2017).
En un sistema CRISPR/Cas9 convencional, la mutación debe de estar ubicada dentro de la región reconocida por el sgRNA, ya que si la secuencia queda intacta, la Cas9 puede reconocer otra vez el mismo sitio del corte después de la reparación por HR e inducir un corte de doble cadena. Este nuevo corte estimularía a la vía de NHEJ y generaría inserciones o deleciones de nucleótidos (indeles) innecesarios. Una nickasa Cas9 evita el reconocimiento del mismo sitio del corte por la nCas9 después de la recombinación homóloga, debido a que el nick en sí mismo se repara por HR sin inducir a un corte de doble cadena y, por lo tanto, no se generan indeles por la vía de NHEJ (Satomura et al., 2017). Un aspecto importante de este sistema de edición es que debe de llevarse a cabo en condiciones en las que se promueva la proliferación celular, ya que la HR es estimulada durante las fases S y G2 del ciclo celular (Branzei & Foiani, 2008).
Para el caso de la edición genética de levaduras no convencionales, las nickasas se han fusionado con editores de bases como las citidina desaminasas (CDA), enzimas que catalizan la desaminación de la citosina y las convierten en uracilo. Es decir, la CDA primero convierte un par C:G en un par U:G. Después de una ronda de replicación o reparación del DNA, el uracilo, al ser complementario a la adenina, y la adenina a la timina, provoca una mutación C→T y el resultado es un par T:A en donde antes había un par C:G (Komor, Kim, Packer, Zuris & Liu, 2016). Sin embargo, en las células, la presencia de un par U:G estimula a las uracilo DNA glucosilasas (UDG), que eliminan al uracilo, y subsecuentemente, reclutan a la vía de reparación por escisión de bases (BER), que restaura al par C:G. Por lo tanto, Komor y colaboradores (2016) integraron un inhibidor de la UDG en este sistema de edición para proteger al par U:G. El papel de la nCas9 es aumentar aún más la proporción de mutaciones C→T. Un nick al lado de un apareamiento erróneo activa a la vía de reparación de los errores de emparejamiento (mismatch repair, MMR), lo que favorece la re-síntesis de la cadena con el nick y que, en este caso, es la que contiene a la guanina del par U:G. Es decir, el nick favorece la reparación del par U:G como un par U:A, en lugar de como un par C:G, posibilitando la obtención de un par T:A después de una ronda de replicación (Komor et al., 2016). Este sistema fue implementado en la levadura S. cerevisiae por Nishida y colaboradores (2016) y nombraron a este sistema Target-AID (targeted activation-induced cytidine deaminase) (Nishida et al., 2016) . Su uso se extendió a la edición genética de las levaduras no convencionales como Kluyveromyces marxianus y Y. lipolytica (Bae, Park, Kim & Hahn, 2020; Nambu-Nishida, Nishida, Hasunuma & Kondo, 2017). En K. marxianus, el sistema Target- AID se utilizó para introducir mutaciones sin sentido (vía una mutación C→T) en dos genes involucrados en la vía de reparación por NHEJ (NEJ1 y DNL4), aumentando la proporción de los eventos de reparación por HR (Nambu-Nishida et al., 2017). En Y. lipolytica, se introdujo una mutación sin sentido en el gen TRP1, involucrado en la biosíntesis del triptófano (Bae et al., 2020).
El sistema CRISPR Nickase es una herramienta que disminuye la citotoxicidad y la generación de indeles al favorecer la reparación por HR sobre NHEJ. La eficiencia de la mutagénesis es elevada en condiciones de alta proliferación celular, además de que las regiones editables son más extensas en comparación con otros sistemas de CRISPR/Cas. Acoplado con CDA e inhibidores de UDG, se ha utilizado con éxito para introducir mutaciones puntuales de C→T de manera muy específica, tanto en levaduras convencionales como en levaduras no convencionales.
Conclusiones
El descubrimiento e implementación de CRISPR/Cas como una tecnología de edición genética y sus variantes, son ejemplos del constante incremento de las estrategias y las herramientas de modificación genética de microorganismos como las levaduras, que son de gran interés para la academia y la industria biotecnológica. CRISPR es actualmente uno de los sistemas de edición genética más diversos, que ofrece varias ventajas, entre ellas la disponibilidad de los diferentes plásmidos y estrategias de expresión, pero también cuenta con algunas limitaciones que no garantizan el funcionamiento de las estrategias creadas para otras especies, por lo que requiere de una optimización en cada modelo de estudio. La mayoría de los intentos exitosos preceden de una comprensión del funcionamiento del sistema CRISPR/Cas in vivo y de la diversidad existente en los procariotas, así como del conocimiento genético de la especie que se desea editar, su vía de reparación del DNA y de la cantidad de sitios PAM disponibles en su genoma. La investigación previa, aunada a la secuenciación de nueva generación, hace relativamente fácil diseñar e implementar CRISPR/Cas de maneras creativas, conjuntas y desafiantes. El desarrollo acelerado de estas herramientas es un salto emocionante para la biotecnología de levaduras no convencionales que han sido poco investigadas y permitirán continuar la exploración de su potencial para convertirlas en especies modelo por sus atributos.

