











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkLista de abreviaturas
LECA, último ancestro común eucarionte; RNAt, ácido ribonucleico de transferencia; RNAr, ácido ribonucleico ribosomal; RNAm, ácido ribonucleico mensajero; TOM, translocasa de la membrana externa; SAM, maquinaria de distribución y ensamblaje; VDAC, canal aniónico dependiente del voltaje; ERMES, estructuras de encuentro entre el retículo endoplásmico y la mitocondria; MIM, maquinaria de importación mitocondrial; TIM22, translocasa de la membrana interna mitocondrial 22; TIM23, translocasa de la membrana interna mitocondrial 23; PAM, motor asociado a la translocasa de presecuencias; MTS, secuencia de localización mitocondrial; MPP, peptidasa de procesamiento mitocondrial; UPRam, respuesta a proteínas desplegadas activada por localización incorrecta de proteínas; mtDNA, DNA mitocondrial; mPOS, estrés provocado por sobreacumulación de precursores mitocondriales; mTAD, degradación de proteínas mitocondriales asociada a la translocación; mitoCPR, respuesta a problemas de importación de proteínas a la mitocondria; ER-SURF, ruta de recuperación superficial del retículo endoplásmico; DCMA, cardiomiopatía dilatada con ataxia; SMDMDM, displasia espondilo metafísea de Megarbane-Dagher-Melki; DDS, síndrome de distonía y sordera; VAV, vectores adenovirales; SDO, sulfuro dioxigenasa; MNGIE, encefalopatía mitocondrial neuro-gastrointestinal; TALENS, nucleasas efectoras similares a activadores transcripcionales; OXPHOS, fosforilación oxidativa.
Introducción
Las mitocondrias son organelos esenciales para la gran mayoría de las células eucariontes. Están compuestas por dos membranas lipídicas, que a su vez delimitan dos espacios acuosos independientes: la matriz y el espacio intermembranal. La membrana interna mitocondrial, además, se pliega hacia el interior de la matriz formando estructuras que se conocen como crestas mitocondriales. La función más estudiada de estos organelos es la producción de energía a través de la fosforilación oxidativa, sin embargo, también llevan a cabo el metabolismo de los aminoácidos y de los lípidos (la beta oxidación) y la formación de centros hierro-azufre. Además, las mitocondrias están estrechamente relacionadas con la apoptosis y, por lo tanto, son uno de los elementos claves de la supervivencia celular.
Origen evolutivo de las mitocondrias
Se ha calculado que el origen de las células eucariontes data de entre 1,000 y 1,900 millones de años (Eme, Sharpe, Brown & Roger, 2014). El surgimiento de estas células ha sido tema de numerosos debates científicos, en particular sobre el tipo de organismos que se asociaron y las presiones evolutivas que favorecieron este proceso. Durante mucho tiempo se pensó que las mitocondrias se originaron por la endosimbiosis de una α-proteobacteria con un protoeucarionte como lo propuso originalmente Lynn Margulis (Sagan, 1993). Sin embargo, hipótesis subsecuentes propusieron que el hospedero de la α-proteobacteria podría haber sido un integrante del dominio Archea (Martin & Müller, 1998). Datos filogenéticos recientes sugieren fuertemente que el hospedero de esta endosimbiosis pertenecía a un arqueón del clado Asgard y que estos procariontes actuales son los familiares más cercanos a los eucariontes (Figura 1, lado izquierdo; Tria et al., 2021; Williams, Cox, Foster, Szöllősi & Embley 2020; Zaremba-Niedzwiedzka et al., 2017). Es posible que el hospedero original no haya sido un arqueón simple, sino un organismo quimérico con gran diversidad genética resultado de interacciones endosimbióticas transitorias anteriores o de eventos masivos de transferencia lateral de genes. En los últimos años, se han postulado diversas hipótesis que explican el establecimiento de la integración física de dos o más células en una y, por lo tanto, el origen de las células eucariontes. En general, en todos los modelos postulados se considera que las interacciones metabólicas entre las células procariontes involucradas favorecieron su asociación permanente con el hospedero. Sin embargo, además de los modelos clásicos de endosimbiosis, actualmente se han planteado mecanismos de fagocitosis o incluso de formación de protuberancias "de adentro hacia afuera" a través de las cuales se explica con mayor parsimonia la formación de la membrana nuclear y del sistema de endomembranas (Figura 1, lado derecho; Baum & Baum, 2014; Eme, Spang, Lombard, Stairs & Ettema, 2017; López-García & Moreira, 2020; Zaremba-Niedzwiedzka et al., 2017). Como se puede apreciar, aún existen grandes incógnitas sobre el origen de las células eucariontes, algunas de ellas se podrán resolver tal vez al obtener mayor información sobre el grupo de las Asgard arqueas, tanto sobre la diversidad dentro de este grupo como de los mecanismos que les permiten establecer relaciones físicas con otros procariontes.
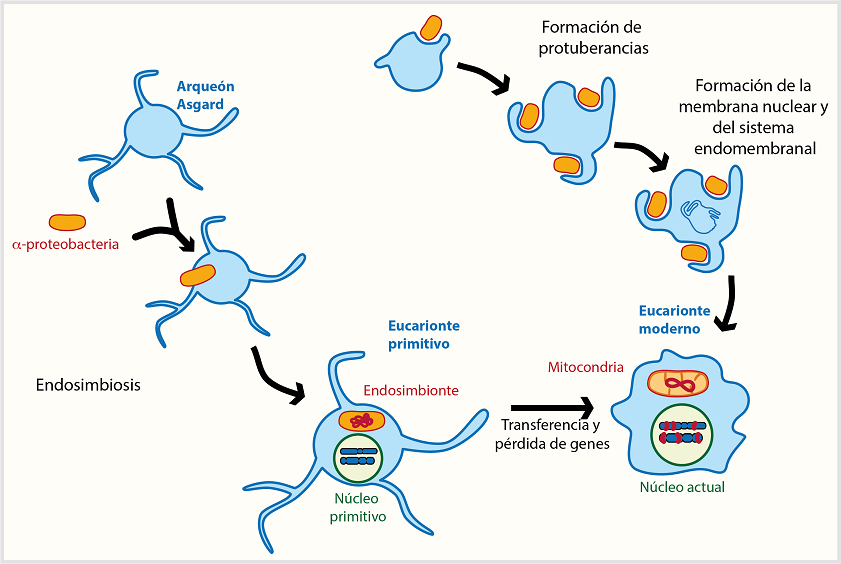
Figura 1 Origen de la célula eucarionte. Existen dos modelos que explican el origen de las células eucariontes: el primero, del lado izquierdo, es la endosimbiosis; propone que el ancestro de un arqueón Asgard engulló una α-proteobacteria para dar lugar a las células eucariontes actuales; el segundo, del lado derecho, indica que el ancestro eucarionte internalizó, mediante protuberancias de su membrana, la α-proteobacteria. Sin importar el modelo, la transferencia de los genes desde la α-proteobacteria, actual mitocondria, al núcleo del hospedero, fue un proceso consecuente. Elaboración personal.
Independientemente de la identidad del hospedero o de su grado de complejidad estructural (por ejemplo, la existencia de otros organelos membranales), esta asociación endosimbiótica dio origen a un ancestro del que se propone derivan todos los eucariontes actuales y que se conoce como el último ancestro común eucarionte o LECA (Last Eukaryotic Common Ancestor, por sus siglas en inglés, Eme et al., 2017; Gray, 2012).
Uno de los desafíos más grandes con el establecimiento de LECA, además de coexistir y organizar el desarrollo celular de dos entidades independientes para asegurar la herencia de esta relación, fue la conservación, la transmisión, la coordinación y la lectura de dos genomas diferentes. Estos dos genomas son el del hospedero que se convirtió en el genoma nuclear y el del huésped que se convirtió en el genoma mitocondrial (Gray, 2015; Karakaidos & Rampias, 2020). Posteriormente, ocurrió una migración del material genético del huésped hacia el genoma del hospedero. Durante esta transferencia ha ocurrido un proceso de continua coevolución entre ambos genomas: a) se perdieron genes irrelevantes para el metabolismo del huésped en su nueva situación de organelo; b) se eliminaron genes con información redundante como procesos metabólicos repetidos y c) se establecieron sistemas bidireccionales de comunicación entre el núcleo y la mitocondria que coordinan la expresión genética de ambos genomas de acuerdo con las necesidades metabólicas de la célula (Bock, 2017). Actualmente, no se conoce totalmente cómo ocurre el proceso de migración de los genes mitocondriales al núcleo; sin embargo, se sabe que de forma natural el mtDNA puede escapar del organelo y convertirse en material genético nuclear o ser usado para la reparación de roturas del DNA de cadena doble en el núcleo (Bock, 2017; Portugez, Martin & Hazkani-Covo, 2018). Existen algunos estudios en levaduras donde se usan marcadores de selección que son transformados en la mitocondria y, de esta forma, es posible calcular la tasa de migración al núcleo, luego de colocar a las levaduras en un medio selectivo; con esta aproximación se ha podido estimar que para los genes mitocondriales la tasa es de 2x10-5, sugiriendo que este proceso continúa ocurriendo (Thorsness & Fox, 1990). Sin embargo, existen genes que son retenidos en el genoma mitocondrial, particularmente aquellos que codifican para proteínas hidrofóbicas, aparentemente, porque representa una desventaja energética importar este tipo de proteínas desde el citosol (Björkholm, Harish, Hagström, Ernst & Andersson, 2015).
Los genomas mitocondriales actuales tienen estructuras muy diversas, algunos son lineales y otros circulares, algunos contienen intrones y otros no, pero todos se componen de un número muy reducido de genes que generalmente codifican para proteínas hidrofóbicas de la cadena respiratoria, RNAt y RNAr (Burger, Gray & Franz Lang, 2003; Ghiselli et al., 2021). Estos genes codifican tan solo el 1% del proteoma mitocondrial, lo que implica que el 99% restante se encuentra codificado en el genoma nuclear (Song, Hermann & Becker, et al., 2021). Esta cantidad masiva de genes codificados en el núcleo son transcritos y posteriormente traducidos por ribosomas citosólicos. Por lo tanto, una necesidad adicional de la endosimbiosis fue el establecimiento de sistemas de reconocimiento y transporte de proteínas desde el citosol hasta la mitocondria (Herrmann, 2003). Es decir, establecer en las proteínas cuyo destino funcional es la mitocondria, una secuencia señal que sea reconocida tanto por las chaperonas en el citoplasma como por los receptores en el organelo, que aseguren su correcta entrega. En consecuencia, las proteínas mitocondriales son sintetizadas en el citosol como proteínas precursoras (también conocidas como proteínas sustrato o proteínas cliente) que únicamente alcanzan su plegamiento funcional al ser localizadas en su destino final (Becker, Song & Pfanner, 2019; Schmidt, Pfanner & Meisinger, 2010). El objetivo de este artículo es: (a) describir las maquinarias que se encargan del transporte de las proteínas mitocondriales y el tipo de secuencia señal que es reconocida por cada una de estas maquinarias (b) detallar las respuestas celulares que se activan como resultado de las alteraciones de estos procesos de distribución; y (c) mencionar las enfermedades ocasionadas por mutaciones en los componentes de importación mitocondrial.
Maquinarias encargadas de la distribución y ensamblaje de las proteínas mitocondriales
El portal de entrada a la mitocondria TOM
El complejo TOM (Translocase of the Outer Membrane, por sus siglas en inglés), localizado en la membrana externa mitocondrial, es el portal de entrada para casi todas las proteínas mitocondriales. TOM, está formado por tres receptores: Tom70, Tom20 y Tom22; tres subunidades pequeñas: Tom5, Tom6 y Tom7, que juegan un rol importante durante la biogénesis y estabilidad del complejo, y Tom40, una proteína con estructura de barril-β que forma el poro por donde las proteínas mitocondriales son translocadas al interior del organelo (Figura 2, verde claro; Araiso et al., 2019; Tucker & Park, 2019).

Figura 2 Rutas de transporte de las proteínas mitocondriales. Una vez que los precursores mitocondriales llegan hasta la periferia mitocondrial asociados a chaperonas, siguen diferentes rutas dependiendo del compartimento mitocondrial donde realizan su función. Las proteínas de la membrana externa mitocondrial tipo barril-β, atraviesan el complejo TOM y son recibidas por las pequeñas Tims que las dirigen al complejo SAM (flechas rosas). Las proteínas de la membrana externa mitocondrial, se anclan a la membrana directamente o utilizan el complejo MIM (flechas verde oscuro). Las proteínas del espacio intermembranal requieren de la proteína Mia40 para su importación (flechas moradas). Los precursores de la membrana interna mitocondrial que son transportadores de metabolitos usan el complejo TIM22 para su inserción, después de pasar por el complejo TOM (flechas azul claro). El complejo TIM23 participa en la importación de proteínas de la matriz mitocondrial (para esto se asocia con PAM) y de la membrana interna mitocondrial (flechas rojas). Finalmente, las proteínas sintetizadas en la mitocondria, son insertadas en la membrana interna mitocondrial por Oxa1. En el cuadro inferior se esquematizan las subunidades que constituyen cada uno de los complejos, sin embargo, los esquemas son sólo una representación gráfica. Los detalles estructurales se amplían en el texto y las Referencias. Elaboración personal.
Los receptores de TOM están anclados en la membrana externa mitocondrial y exponen los dominios de unión a sustratos hacia el citosol. Estos receptores reciben a las proteínas mitocondriales mediante el reconocimiento de diversas secuencias señal. Mientras que Tom20 reconoce señales que dirigen proteínas hasta la matriz mitocondrial, Tom70 reconoce señales hidrofóbicas de proteínas cuyo destino es la membrana interna mitocondrial (Backes et al., 2018; Brix, Dietmeier & Pfanner, 1997; Brix, Rudiger, Bukau, Scheneider-Mergener & Pfanner, 1999; Yamamoto et al., 2009; Yamano et al., 2008). Una vez que la proteína sustrato es reconocida por Tom20 o Tom70, esta es transferida al tercer receptor Tom22, cuyo papel es entregar a las proteínas al poro de translocación Tom40. Los sustratos cruzan la membrana externa mitocondrial a través de Tom40 y son recibidas en el espacio intermembranal por el extremo C-terminal de Tom22 (Mani, Rout, Desy & Schneider, 2017; Shiota, Mabuchi, Tanaka-Yamano, Yamano & Endo, 2011). De aquí en adelante, la proteína sustrato es entregada a otros complejos dentro de la mitocondria para su correcta distribución hacia el subcompartimento mitocondrial que será su destino final.
Recientemente, se han realizado esfuerzos por esclarecer la estructura y la estequiometría del complejo TOM en Saccharomyces cerevisiae y Neurospora crassa, así como en Homo sapiens (Araiso et al., 2019; Bausewein et al., 2017; Tucker & Park, 2019; Wang et al., 2020). De manera general, el complejo TOM es un dímero formado por dos poros Tom40 que están unidos por dos subunidades Tom22, las subunidades pequeñas Tom5, Tom6 y Tom7 se ubican en la periferia del poro de Tom40 de forma que Tom6 y Tom7 se encuentran en posiciones opuestas. Asimismo, se plantea la posibilidad de algunos estados de oligomerización del complejo TOM, al sugerir una transición de dímero a trímero (Araiso et al., 2019; Shiota et al., 2015) o incluso la formación de un tetrámero (Bausewein et al., 2017; Tucker & Park, 2019; Wang et al., 2020). Ninguna de las estructuras reportadas contiene a los receptores Tom20 y Tom70; por lo que, una de las preguntas abiertas más importantes en cuanto a la organización de este complejo es si la asociación de estas dos subunidades es transitoria, o si existen subtipos de complejos TOM, según la clase de precursores mitocondriales a importar.
Pequeñas Tims, chaperonas del espacio intermembranal independientes de ATP
Las pequeñas Tims (pTims) son chaperonas localizadas en el espacio intermembranal que se unen a los precursores mitocondriales, evitando así que sus residuos hidrofóbicos queden expuestos al ambiente hidrofílico (Petrakis, Alcock & Tokatlidis, 2009; Wiedemann, Pfanner & Chacinska, 2006). Adicionalmente, las pTims participan en dirigir a estos precursores hacia la membrana externa o interna mitocondriales, donde los complejos proteicos SAM y TIM22, respectivamente, los insertan y ensamblan. Entre los sustratos de las pTims están: i) los transportadores de metabolitos como el transportador ADP/ATP (Curran, Leuenbeger, Oppliger & Koehler, 2002; Endres, Neupert & Brunner, 1999; Koehler et al., 1998; Sirrenberg et al.,, 1998; Vasiljev et al., 2004); ii) las proteínas tipo barril-β como Tom40 (Wiedemann et al., 2004); y iii) la proteína Tim23, una subunidad del complejo TIM23 (Curran, Leuenberger, Oppliger & Koehler, 2002; Davis, Alder, Jensen & Johnson, 2007; Davis, Sepuri, Holder, Johnson & Jensen, 2000).
Las pTims son: Tim8, Tim9, Tim10, Tim12 y Tim13, las cuales forman complejos hexaméricos alternados. Aunque el complejo funcional predominante es Tim93-Tim103 que es esencial en la levadura S. cerevisiae (Figura 2, hexámero verde y rosa; Vergnolle et al., 2005; Webb, Gorman, Lazarou, Ryan & Gulbis, 2006); existe también el complejo análogo Tim83-Tim133. Por otro lado, Tim12 forma un complejo con Tim93-Tim103, relevante para la unión con el complejo TIM22, que será mencionado más adelante (Koehler et al.,, 1998; Sirrenberg et al., 1998). Los complejos Tim93-Tim103 y Tim83-Tim133 pesan aproximadamente 60 kDa y su estructura está formada por a-hélices cuya forma se asemeja a una propela de 6 palas, donde cada una de las palas es una proteína Tim (Baker et al., 2009; Beverly, Sawaya, Schmid & Koehler, 2008; Webb et al., 2006). Cada pTim tiene un motivo de cisteínas CX3CXnCX3C (donde 11>n<16), importante en la formación de dos puentes disulfuro relevantes para su función y su estructura (Koehler, 2004).
Las proteínas con estructura barril-β utilizan la vía de inserción mediada por SAM
Una particularidad de la mitocondria es la presencia de proteínas con estructura de barril-β en la membrana externa mitocondrial. Este tipo de proteínas también están presentes en las bacterias Gram-negativas y en la membrana externa de los cloroplastos (Chaturvedi & Mahalakshmi, 2017). Algunos ejemplos de barriles-β mitocondriales son el poro de translocación Tom40, la porina (también llamada VDAC), que es una proteína que permite el transporte de iones y metabolitos, la subunidad Mdm10 del complejo ERMES, involucrado en mediar contactos mitocondria-retículo endoplásmico (Fairman, Noinaj & Buchanan, 2011; Flinner et al., 2013; Tucker & Park, 2019) y la subunidad Sam50 del complejo SAM, cuyo rol es la inserción de barriles-β en la mitocondria.
Insertar y plegar barriles-β en la membrana externa mitocondrial es un proceso costoso desde el punto de vista energético. El sistema de transporte de estos sustratos consta de tres pasos sucesivos que involucran a tres diferentes complejos proteicos (Figura 2, flechas rosas). En el primer paso, el precursor del barril-β llega al complejo TOM para ingresar a la mitocondria. El segundo paso, ocurre cuando el sustrato alcanza el espacio intermembranal y se asocia al complejo Tim93-Tim103 (Habib, Waizenegger, Lech, Neupert & Rapaport, 2005; Weinhäupl et al., 2018). Finalmente, el tercer paso implica la entrega al complejo SAM del sustrato (Sorting and Assembly Machinery, por sus siglas en inglés), en la membrana externa mitocondrial (Gentle, Gabriel, Beech, Waller & Lithgow, 2004; Qiu et al.,, 2013; Wenz et al., 2015; Wiedemann et al., 2003).
SAM está formado por tres subunidades, Sam35, Sam37 y Sam50. La subunidad Sam50 es el núcleo del complejo y adopta una estructura barril-β (Kozjak et al., 2003; Stroud et al., 2011). El modelo actual establece que Sam50 utiliza su propia estructura para facilitar el plegamiento de otros barriles-β (Hohr et al., 2018; Noinaj et al., 2013). Sam35 y Sam37 son subunidades periféricas de SAM que se anclan a la membrana externa mitocondrial y quedan expuestas hacia el citosol. Sam35 funciona como receptor para los sustratos barril-β, mediante el reconocimiento de las señales-β en su extremo C-terminal (Figura 3, caja rosa; Chan & Lithgow, 2008; Habib et al., 2007; Milenkovic et al., 2004). Por su parte, Sam37 promueve la liberación de los barriles-β en la membrana externa mitocondrial una vez que han sido plegados correctamente (Chan & Lithgow, 2008). Adicionalmente, Sam37 promueve la asociación transitoria que ocurre entre TOM y SAM durante la biogénesis de los barriles-β (Qiu et al., 2013; Wenz et al., 2015). Esta asociación es una estrategia que minimiza el tiempo de exposición de los sustratos hidrofóbicos en el medio acuoso del espacio intermembranal. Finalmente, se ha sugerido que Sam37 podría funcionar, de manera similar a como lo hace Tom70, como un receptor para proteínas mitocondriales que deben ser translocadas al interior del organelo (Gratzer et al.,, 1995; Ponce-Rojas et al., 2017). La eliminación simultánea del gen SAM37 con TOM70 genera un fenotipo letal en la levadura S. cerevisiae, lo cual sugiere que los dos receptores tienen papeles funcionales al menos, parcialmente redundantes. Sin embargo, también se ha observado que la asociación de Sam37 a TOM modifica ligeramente la afinidad de Tom70 por sus sustratos (Gratzer et al., 1995). Por lo que es necesario realizar estudios adicionales para determinar hasta qué punto las funciones de Sam37 y Tom70 se sobrelapan, así como para determinar cómo la acción de uno modifica la especificidad del otro, durante el reconocimiento de las proteínas mitocondriales.
El complejo MIM inserta proteínas con cruces trans-membranales a la membrana externa mitocondrial
Además de los barriles-β, existen proteínas mitocondriales con topología distinta en la membrana externa mitocondrial, ancladas por medio de uno o varios cruces transmembranales. Los cruces pueden localizarse tanto en el extremo amino como en el carboxilo terminal de las proteínas. Recientemente, se ha descrito que el complejo MIM (Mitochondrial Import Machinery, por sus siglas en inglés) promueve la inserción de este tipo de proteínas (Figura 2, flecha verde oscuro; Becker et al., 2011; Papic, Krumpe, Dukanovic, Dimmer & Rapaport, 2011). El complejo MIM, localizado en la membrana externa mitocondrial, está formado por múltiples copias de la proteína de un cruce transmembranal Mim1 que se asocian formando un poro de translocación, además de una o dos copias de la proteína Mim2 (Dimmer et al., 2012; Kruger et al., 2017). MIM funciona en tres modos distintos: i) en forma individual, participa en la inserción de proteínas que tienen un cruce transmembranal (Doan et al., 2020); ii) asociado al receptor Tom70, facilitando así la inserción de proteínas con uno o varios cruces transmembranales; o iii) acoplado al complejo SAM, para en conjunto insertar a las subunidades pequeñas del complejo TOM y sus receptores Tom20 y Tom70 en la membrana externa mitocondrial (Becker et al., 2008; Popov-Celeketić, Waizenegger & Rapaport, 2008). Es posible que en este último caso la participación de MIM con SAM también asegure un ensamblaje eficiente y coordinado del complejo TOM. Esta coordinación durante el ensamblaje del complejo TOM podría evitar que sus subunidades individuales se encuentren en puntos distantes dentro de la membrana externa mitocondrial y de esta manera, afecten la importación de proteínas. Otra pregunta abierta es si la proteína Tom40 puede actuar sin las otras subunidades del complejo TOM, lo que podría provocar un paso desregulado de proteínas entre el citosol y el espacio intermembranal.
El sistema de relevo oxidativo Mia40-Erv1 retiene proteínas en el espacio intermembranal y ayuda a su plegamiento
La oxidorreductasa Mia40 es una proteína anclada en la membrana interna mitocondrial en la levadura S. cerevisiae, pero en las plantas y en los mamíferos se trata de una proteína soluble (CHCHD4 en mamíferos, Backes & Herrmann, 2017). Todos los homólogos de Mia40 comparten una región central en la que hay seis residuos de cisteína invariables organizados en un motivo CPC y dos motivos CX9C (donde X puede ser cualquier aminoácido). Mia40 recibe en el espacio intermembranal a las proteínas que ya cruzaron la membrana externa mitocondrial y establece enlaces disulfuro transitorios con las proteínas entrantes para evitar su regreso al citosol. Mia40 funciona también como chaperona al facilitar el plegamiento oxidativo de sus proteínas sustrato, ya que favorece el establecimiento de puentes disulfuro intramoleculares o la unión con iones divalentes (Figura 2, flechas púrpura; Backes & Herrmann, 2017; Stojanovski, Bragoszewski & Chacinska, 2012).
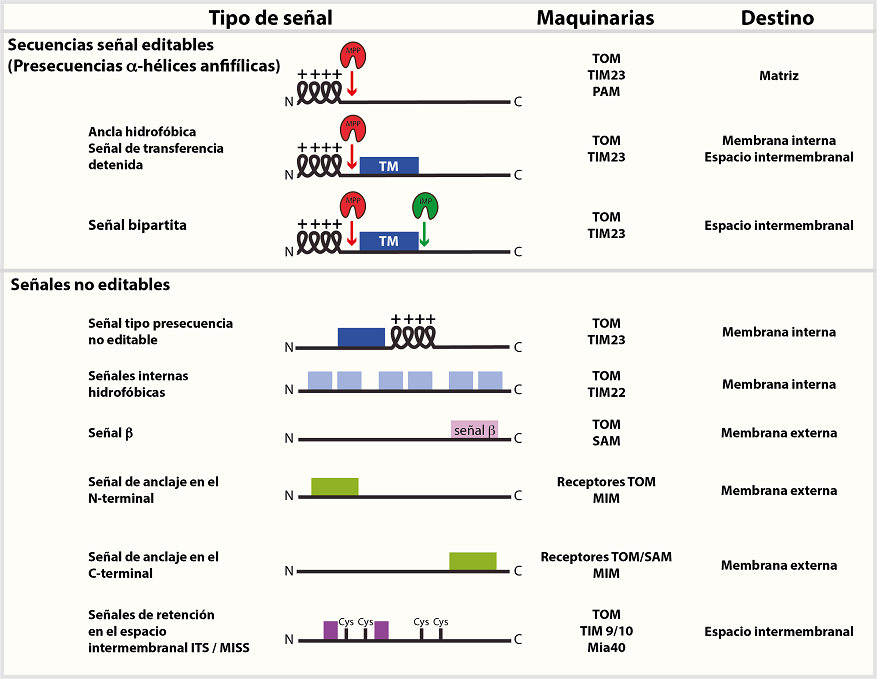
Las proteínas sustrato de Mia40 contienen residuos de cisteína en motivos CX3C o CX9C que son importantes, por un lado, para su retención en el espacio intermembranal, puesto que muchas de estas proteínas tienen una masa molecular pequeña y pueden entrar o salir de la mitocondria a través del complejo TOM (Stojanovski et al., 2012); y, por otro lado, para los pasos posteriores de plegamiento oxidativo. Los sustratos contienen una secuencia señal de localización en el espacio intermembranal conocida como ITS o MISS, que se compone de 9 aminoácidos río arriba o río abajo de la cisteína importante para el anclaje. Esta secuencia señal forma una hélice anfipática que expone residuos hidrofóbicos que se acoplan a una hendidura hidrofóbica en Mia40 (Figura 3; Milenkovic et al., 2009; Sideris et al., 2009).
Una vez que llega al espacio intermembranal, la proteína cliente forma un puente disulfuro transitorio con Mia40; paso inicial en el relevo redox entre ambas proteínas. Posteriormente, Mia40 se asocia tanto con el sustrato como con la proteína Erv1 para transferir los electrones liberados con la formación de los puentes disulfuro y permitir así la oxidación del precursor. Una vez oxidados, los sustratos son liberados en el espacio intermembranal y Mia40 queda en estado reducido. Para que un nuevo ciclo de importación comience, es necesario que Mia40 regrese a su estado oxidado. Esto se logra gracias a Erv1, que acepta los electrones de Mia40 reducida a través de la formación de nuevos puentes disulfuro. Finalmente, Erv1 transfiere los electrones a través del citocromo c a la cadena respiratoria, y, por lo tanto, al oxígeno, quien es el aceptor final de los electrones en el proceso. Es importante mencionar que la presencia del glutatión en el espacio intermembranal es necesaria para crear un ambiente que favorezca la formación de los puentes disulfuro (Bien et al., 2010).
Los transportadores de metabolitos se insertan mediante TIM22 en la membrana interna mitocondrial
TIM22 (Translocase of the Inner Membrane, por sus siglas en inglés) es un complejo presente en la membrana interna mitocondrial encargado de insertar y ensamblar a los transportadores de metabolitos, en este mismo compartimento mitocondrial (Endres et al., 1999; Gomkale et al., 2020; Kubrich, Rassow, Voos, Pfanner & Honlinger, 1998; Rampelt et al., 2020; Sirrenberg, Bauer, Guiard, Neupert & Brunner, 1996). El complejo TIM22 está formado por tres subunidades: Tim22, la cual forma el poro de translocación (Kubrich et al., 1998; Sirrenberg et al., 1996); Tim54, el receptor del complejo (Kerscher, Holder, Srinivasan, Leung & Jensen, 1997) y Tim18 que, además de mediar la inserción de las proteínas en la membrana interna mitocondrial, también promueve el ensamblaje y da estabilidad al complejo (Kerscher, Sepuri & Jensen, 2000; Koehler et al., 2000). Se ha observado que la proteína succinato deshidrogenasa 3 (Sdh3) interacciona con Tim18 y favorece el ensamblaje del complejo TIM22 (Gebert et al., 2011).
Los complejos TOM y TIM22 trabajan en forma conjunta en cinco pasos que se describen a continuación (Figura 2, flechas lavanda; Pfanner & Neupert, 1987; Rehling, Brandner & Pfanner, 2004): i) el precursor mitocondrial es llevado hasta el complejo TOM por chaperonas presentes en el citosol (Young, Hoogenraad & Hartl, 2003) donde ii) interacciona con el receptor Tom70, que reconoce las secuencias internas en los precursores (Brix et al., 2000; Wiedemann, Pfanner & Ryan, 2001; Young et al., 2003), iii) al cruzar la membrana externa mitocondrial, el sustrato es recibido por las chaperonas Tim93-Tim103 que lo entregan al complejo TIM22 (Koehler et al., 1998; Murphy, Leuenberger, Curran, Oppliger & Koehler, 2001), en este paso es importante la participación de la chaperona Tim12 (Koehler et al., 1998; Sirrenberg et al., 1996), ya que ésta facilita que Tim54 reconozca a la proteína sustrato, iv) el precursor es insertado y ensamblado en el poro del complejo aunque este proceso no se ha estudiado con gran detalle (Rehling et al., 2004) ; y finalmente v) la proteína es liberada lateralmente e insertada en la membrana interna mitocondrial.
Las proteínas que son clientes del complejo TIM22, también presentan señales de localización mitocondrial caracterizadas por 6 cruces transmembranales de aproximadamente 10 aminoácidos cada uno; estas señales están integradas a la proteína y no son editables (Figura 3; Chacinska, Koehler, Milenkovic, Lithgow & Pfanner, 2009).
Las proteínas cuyo destino es la matriz mitocondrial utilizan la ruta TIM23-PAM
El segundo complejo translocador de proteínas situado en la membrana interna mitocondrial es TIM23 (Figura 2, azul; Translocase of the Inner Membrane, por sus siglas en inglés), quien se encarga de facilitar la translocación de las proteínas que residen en la matriz mitocondrial o de insertarlas en la membrana interna, de una manera dependiente del potencial de membrana mitocondrial (ΔΨ; Callegari, Cruz-Zaragoza & Rehling, 2020; Mokranjac & Neupert, 2010; Schulz, Schendzielorz & Rehling, 2015). El potencial de membrana mitocondrial es un gradiente electroquímico que se establece a través de la membrana interna tanto por el paso de protones de la matriz mitocondrial al espacio intermembranal, por los complejos I, III y IV de la cadena respiratoria, así como por el paso de los electrones derivados de las reacciones de óxido-reducción del ciclo de Krebs. El gradiente de protones es aprovechado también para la síntesis de ATP (Zorova et al.,, 2018; Rich & Maréchal, 2010).
TIM23 está formado por las siguientes subunidades: Tim17 y Tim23 que forman el poro de translocación (Dekker, et al., 1993; Kubrich et al., 1994; Maarse, Blom, Keil, Pfanner & Meijer, 1994); Tim50, es el receptor que reconoce las presecuencias de los precursores que provienen del complejo TOM (Geissler et al., 2002; Mokranjac et al., 2003; Mokranjac et al., 2009; Yamamoto et al., 2002) y regula también la permeabilidad del poro formado por Tim23-Tim17 (Meinecke et al., 2006) y Tim21, que es una proteína integral de membrana (Chacinska et al., 2005; Mokranjac, Popov-Celeketić, Hell & Neupert, 2005) capaz de interaccionar con el complejo TOM a través del receptor Tom22 (Albrecht et al., 2006; Chacinska et al., 2005). Además, Tim21 participa en la asociación del complejo TIM23 con los complejos de la cadena respiratoria (van der Laan et al., 2006). Así, al crearse un potencial de membrana localizado, se facilita la translocación electroforética de las presecuencias y, por tanto, del resto de la proteína entrante.
Los precursores que se dirigen hacia la matriz mitocondrial contienen una presecuencia y utilizan la llamada "ruta de la presecuencia" (en inglés Presequence Pathway), que requiere el acoplamiento de TIM23 con el complejo motor PAM (Figura 2, azul cielo; Presequence translocase-Associated Motor, por sus siglas en inglés; Berthold et al., 1995; Mokranjac, 2020). Esta presecuencia llamada MTS (Matrix Targeting Sequence, por sus siglas en inglés; Figura 3) está en el extremo N-terminal de las proteínas y se caracteriza por ser una alfa-hélice anfipática, con una cara cargada positivamente y la otra con aminoácidos hidrofóbicos; es de longitud variable, aunque generalmente está formada por 20-50 aminoácidos (von Heijne, 1986).
La función de PAM es promover la translocación de las proteínas sustrato a la matriz mitocondrial mediante el consumo de ATP. TIM23 y PAM se asocian a través de la proteína Tim44 (Maarse, Blom, Grivell & Meijer, 1992) que a su vez sirve como andamio para la chaperona dependiente de ATP mtHsp70/Ssc1, el primer componente de PAM (Kang et al., 1990; Kronidou et al., 1994; Rassow et al., 1994; Schneider et al., 1994). mtHsp70 a su vez requiere de las cochaperonas Pam18 (D'Silva, Schilke, Walter, Andrew & Craig, 2003; Truscott et al., 2003) y Pam16 (Frazier et al., 2004) así como del factor de recambio de nucleótidos Mge1 (Miao, Davis & Craig, 1997). De esta forma, cuando un precursor atraviesa el complejo TIM23, se asocia a la mtHsp70 a través del dominio de unión al sustrato, mientras que el dominio de unión a nucleótidos, se une al ATP; acto seguido las proteínas Pam18 y Pam16 favorecen la hidrólisis del ATP y Mge1 intercambia el ADP por ATP, permitiendo la liberación del precursor y así, comienza un nuevo ciclo. Cada uno de estos ciclos de unión-liberación, permite la translocación de las proteínas hacia la matriz mitocondrial. Una vez que el precursor ingresa a la matriz mitocondrial, su MTS es eliminada por la proteasa de la matriz mitocondrial MPP (Mitochondrial Processing Peptidase, por sus siglas en inglés; Hawlitschek et al., 1988), para que el sustrato alcance su estado maduro y adquiera el plegamiento apropiado para su función. Existe otra subunidad accesoria, Pam17, a la que se le ha adjudicado la función de estabilizar la unión de Pam18-Pam16 y de esta orma, promover la asociación con Tim23 y Tim17 (van der Laan et al., 2005).
Como se mencionó anteriormente, las presecuencias dirigen a las proteínas hacia la matriz mitocondrial, sin embargo, pueden acompañarse de otros motivos que son interpretados por TIM23 y que determinan que el destino de un precursor no sea precisamente la matriz mitocondrial. Lo anterior acontece cuando una MTS es seguida de un cruce transmembranal que funciona como un ancla hidrofóbica (Figura 3). En este caso a señal indica que las proteínas deben ser insertadas en la membrana interna mitocondrial en un proceso denominado "transferencia detenida" (en inglés Stop Transfer;Glick et al., 1992; van der Laan et al., 2007). En estos casos, la MTS se expone a la matriz; sin embargo, el cruce transmembranal se retiene en el canal de TIM23 y la proteína es liberada lateralmente con ayuda de la proteína Mgr2 (Ieva et al., 2014; Lee et al., 2020; Matta, Kumar & D'Silva, 2020). Ejemplos de proteínas que siguen esta ruta de importación son los citocromos c1 y b2 (Glick et al., 1992). Una modificación de este proceso permite liberar a la proteína madura directamente al espacio intermembranal, cuando después del cruce transmembranal existe una secuencia de corte que es reconocida por las proteasas de la membrana interna mitocondrial (Gakh, Cavadini & Isaya, 2002). Por tener dos sitios de corte, este tipo de señales se conocen como señales bipartitas (Figura 3) como es el caso de la proteína Smac/Diablo (Burri et al., 2005).
En ocasiones, los sustratos contienen más de un cruce transmembranal y, en estos casos, el segundo cruce es el que indica que se debe detener la importación, para que la proteína pueda ser liberada lateralmente a la membrana. Cuando esto ocurre, el primer cruce llega hasta la matriz mitocondrial y, posteriormente, se inserta en la membrana interna mitocondrial mediante un mecanismo llamado "distribución conservada" (en inglés Conservative Sorting) utilizando a la maquinaria Oxa1 (ver sección siguiente; Hartl, Schmidt, Wachter, Weiss & Neupert, 1986; Hell, Herrmann, Pratje, Neupert & Stuart,1998; Herrmann, Neupert & Stuart, 1997). Es importante considerar que existen proteínas sustratos que utilizan tanto la "distribución conservada" como la "transferencia detenida" para alcanzar su topología final; como sucede para la proteína Mdl1, que ilustra la cooperación que existe entre los diferentes componentes de los sistemas de transporte de proteínas mitocondriales (Bohnert et al., 2010).
Oxa1 inserta en la membrana interna mitocondrial proteínas codificadas en el genoma mitocondrial
La translocasa Oxa1 es un miembro de la familia YidC/Oxa1/Alb3, presente en bacterias, mitocondrias y cloroplastos, respectivamente, por lo que se presume que los integrantes de esta familia están presentes en los organelos que fueron heredados de las bacterias endosimbiontes que les dieron origen. Los tres homólogos tienen una región central conservada de cinco cruces transmembranales esencial para insertar y plegar a las proteínas membranales (Funes, Kauff, van der Sluis, Ott & Herrmann, 2011).
Localizada en la membrana interna mitocondrial, Oxa1 interacciona con proteínas recién importadas por el complejo TIM23 hasta la matriz y facilita su inserción en la membrana interna mitocondrial en una reacción de exportación, es decir, desde la matriz hacia la membrana interna mitocondrial (Figura 2, turquesa; Hell et al., 1998). Además, media la inserción de proteínas codificadas en el genoma mitocondrial, al facilitar la asociación de los ribosomas mitocondriales con la membrana interna mitocondrial, en un proceso denominado inserción cotraduccional, ya que la síntesis de la proteína y la inserción suceden de forma simultánea. Este proceso requiere la presencia de otras proteínas como Mba1, Mdm38 y Mrx15 para asegurar el anclaje del ribosoma con la membrana interna mitocondrial (Bauerschmitt et al., 2010; Möller-Hergt, Carlström, Stephan, Imhof & Ott, 2018; Pfeffer, Woellhaf, Herrmann & Förster, 2015; Preuss, Ott, Funes, Luirink & Herrmann, 2005). La función de Oxa1 repercute indirectamente en la biogénesis de otros sustratos mitocondriales; esto se debe a que Oxa1 es necesaria para el ensamblaje del módulo Tim18-Sdh3 y, por tanto, la ausencia de Oxa1 compromete la inserción en la membrana interna de los transportadores de metabolitos dependientes de TIM22 (Hildenbeutel et al., 2012; Stiller et al., 2016).
Respuestas celulares a fallas en los sistemas de transporte e importación de las proteínas mitocondriales
Dado que la importación de las proteínas a la mitocondria es un proceso complejo que implica diferentes pasos que van desde la síntesis, pasando por el transporte, la importación y la ubicación en el compartimento mitocondrial correspondiente, está sujeto a fallas. Sin embargo, existen respuestas celulares que ayudan a contrarrestar estas alteraciones. En esta sección, se mencionan las principales características de las respuestas celulares activadas cuando ocurren alteraciones en los sistemas de transporte de las proteínas mitocondriales; un campo de investigación muy activo en los últimos años. Es importante mencionar que, en general, estas respuestas implican cambios tanto transcripcionales como traduccionales en la célula; es decir, se activan factores de transcripción que inducen la expresión de los genes relacionados con las respuestas al estrés; al tiempo que disminuye la síntesis de proteínas no requeridas (Boos et al., 2019).
Cualquier alteración en los sistemas de transporte implica la acumulación de precursores mitocondriales en el citosol o en el poro de translocación Tom40. Un ejemplo del primer caso es la UPRam o mPOS (Unfolded Protein Response activated by mistargeting of proteins o mitocondrial Precursor Over-accumulation Stress, por sus siglas en inglés respectivamente; Wang & Chen, 2015; Wrobel et al., 2015), ocasionada por alteraciones en el funcionamiento de Mia40 y cuyo resultado es un incremento en la actividad del proteosoma (la maquinaria encargada de la degradación de las proteínas en el citosol) y una reprogramación traduccional (Figura 4, lado izquierdo, flechas azules claras). En el segundo caso, la respuesta se llama mTAD (Mitochondrial protein Translocation-Associated Degradation, por sus siglas en inglés), caracterizada por la detección de la oclusión del complejo TOM por parte de la proteína Ubx2, que recluta a la AAA-ATPasa Cdc48 y remueve al precursor para su posterior degradación vía el proteosoma (Figura 4, lado izquierdo, flechas negras; Mårtensson et al., 2019). Estudios recientes han mostrado que Ubx2 también favorece el marcaje por ubiquitinación de las proteínas mitocondriales que por aumentos en la temperatura no son funcionales y, por lo tanto, no son importadas a la mitocondria. La función de Ubx2 requiere del reclutamiento de las proteínas Doa1-Ufd1-Npl4. Los sustratos no funcionales son ubiquitinados por Ubx2 una vez que fueron reconocidos por las chaperonas de la familia SSA y Sis1 (Figura 4, lado izquierdo, flechas grises; Metzger, Scales, Dunklebarger, Loncarek & Weissman, 2020).
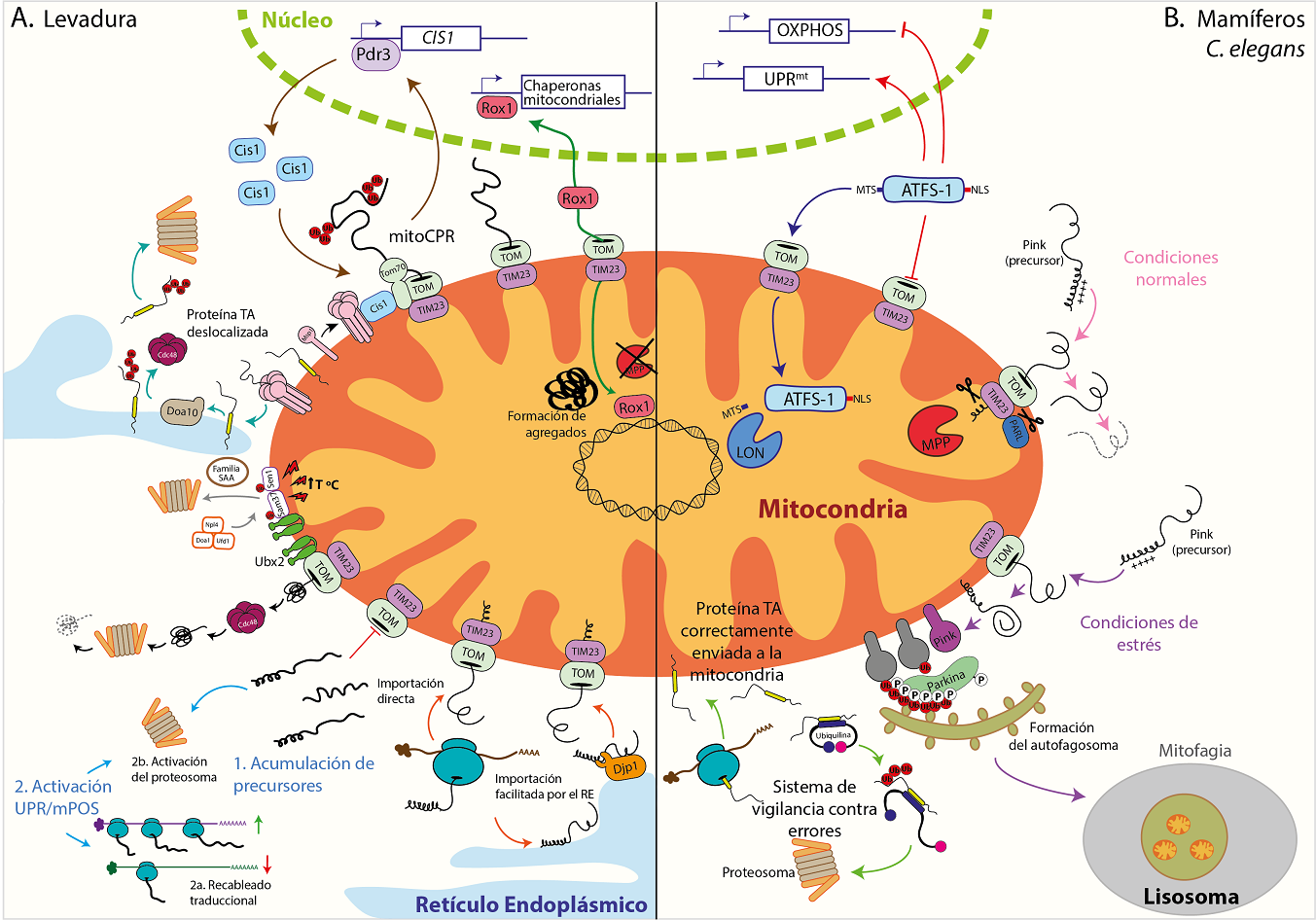
Figura 4 Respuestas celulares a fallas en los sistemas de transporte y disfunción mitocondrial. A. Levadura Sacharomyces cerevisiae Rox1 (rojo fucsia) es un factor de transcripción que al fallar la proteasa MPP (rojo) se dirige al núcleo y promueve la transcripción de las chaperonas mitocondriales (flechas verdes). Fallas en las maquinarias de importación, como la obstrucción de TOM, inician diferentes respuestas: por un lado el reclutamiento de la proteína Mspl, que extrae al precursor para ser degradado (flechas cafés) y por otro la intervención de Cdc48 (rojo vino), reclutada por Ubx2 (verde limón), para extraer el precursor atascado del canal de TOM (flechas negras). La acumulación de precursores en el citosol ocasiona un reprogramado de la traducción para incrementar la síntesis de proteínas del proteosoma a través de UPRam/mPOS (flechas azules claras). Las alteraciones en las proteínas por cambios en la temperatura son detectados por Ubx2 (verde limón), que recluta al sistema Doa1-Ufd1-Npl4 y marca con ubiquitina (rojo cereza) a las proteínas dañadas para su posterior degradación (flechas grises). Las fallas en la distribución de las proteínas, tienen sus sistemas correctivos: las proteínas con topología TA del retículo endoplásmico, pero que se encuentran erróneamente en la mitocondria son re-localizadas por Msp1 (rosado claro) al retículo endoplásmico, donde son ubiquitinadas y retiradas para su degradación (flechas turquesas). Por su parte, las proteínas mitocondriales pueden ser dirigidas de forma errónea al retículo endoplásmico, sin embargo, Djpl (naranja) es capaz de redirigirlas a la mitocondria (flechas naranjas) B. Mamíferos y C. elegans. La respuesta mitocondrial a proteínas mal plegadas o mtUPR, se caracteriza por la localización dual (mitocondrial/nuclear) de ATFS-1 (azul claro): cuando hay fallas en la mitocondria, ATFS-1 se dirige al núcleo para promover la transcripción de genes que ayuden a contrarrestar la falla (flechas azules y rojas). PINK es una proteína mitocondrial que en condiciones de estrés se encuentra en la membrana externa mitocondrial y desde allí dirige una cascada de eventos junto con Parkina (fosforilación y ubiquitinación; verde pistacho), que actúa como señal para reclutar a la maquinaria autofágica (flechas rosadas y moradas). Las ubiquilinas (azul ultramar), son chaperonas citosólicas que reconocen a las proteínas con topología TA que no son dirigidas correctamente a la mitocondria, redirigen a estas proteínas o las marcan para su degradación (flechas verdes limón). Elaboración personal.
Otra alteración en el sistema de importación ocurre cuando la peptidasa MPP no funciona adecuadamente e impide la correcta maduración de los precursores y el inicio de una respuesta temprana a proteínas mal plegadas, ya que se generan agregados proteicos en la matriz mitocondrial. Esta respuesta ocasiona que el factor de transcripción Rox1 sea importado a la mitocondria para unirse al ADN mitocondrial y, de esta forma, se asegure la relación funcional coordinada mitocondria-núcleo (Figura 4, lado izquierdo, flechas verdes; Poveda-Huertes et al., 2020).
Otra proteína capaz de retirar precursores atrapados en Tom40 es Msp1, una AAA-ATPasa presente en la membrana externa mitocondrial. Alteraciones en el potencial electroquímico transmembranal (ΔΨ) pueden afectar la translocación de los precursores a través de Tom40 y bloquearlo. Msp1 es capaz de retirar al precursor que obstruye a Tom40 y dirigirlo a su degradación por el proteosoma (Figura 4, lado izquierdo, flechas cafés; Basch et al., 2020; Weidberg & Amon, 2018). Es importante mencionar que, para el caso de las proteínas con una señal bipartita, el paso de extracción requiere a la proteína Cis1, esta respuesta particular se denomina mitoCPR (mitochondria Compromised Protein-import Response, por sus siglas en inglés; Weidberg & Amon, 2018). El rol de Msp1 va más allá de rescatar a Tom40 de precursores atrapados, también es capaz de identificar proteínas del retículo endoplásmico que han sido insertadas erróneamente en la membrana externa mitocondrial y llevarlas a su destino original, donde son ubiquitinadas y extraídas por Cdc48 para su posterior degradación por el proteosoma (Figura 4, lado izquierdo, flechas turquesas; Dederer et al., 2019; Matsumoto et al., 2019).
Los errores en la distribución de las proteínas hacia los organelos incorrectos abarcan más ejemplos que el mencionado anteriormente. En ocasiones algunas proteínas mitocondriales pueden ser dirigidas erróneamente al retículo endoplásmico; en donde la proteína Djp1 (localizada en la membrana del propio retículo) es capaz de reconocerlas y redirigirlas a la mitocondria para que continúen su proceso de importación y ensamblaje (Figura 4, lado izquierdo, flechas naranjas; Hansen et al.,, 2018). Este mecanismo se denomina ER-SURF (Endoplasmic Reticulum SURFace retrival pathway, por sus siglas en inglés). Otras proteínas, como las ubiquilinas de mamíferos, reconocen proteínas mitocondriales con topología TA (Tail-Anchored, por sus siglas en inglés; proteínas ancladas por un segmento transmembranal en su extremo C-terminal) que no fueron correctamente integradas en la membrana externa mitocondrial; las ubiquilinas son capaces de redirigirlas o marcarlas para su posterior degradación (Figura 4, lado derecho, flechas verde limón; Itakura et al., 2016).
En mamíferos también existen respuestas celulares por daño mitocondrial. El sistema más conocido es el par PINK1/Parkina, proteínas descubiertas en pacientes con enfermedad de Parkinson (Kitada et al., 1998; Valente et al., 2001), una enfermedad neurodegenerativa relacionada fuertemente con disfunción mitocondrial (Abou-Sleiman, Muquit & Wood, 2006). PINK1 es una cinasa que actúa como sensor del daño mitocondrial, generalmente ocasionado por una disminución en el potencial de membrana. Su función es fosforilar ubiquitinas previamente añadidas a proteínas de la membrana externa mitocondrial por Parkina. La acción conjunta de PINK1/Parkina genera ciclos de ubiquitinación-fosforilación en algunas proteínas de la membrana externa mitocondrial; esto ciclos funcionan como señal para el reclutamiento de la maquinaria autofágica y llevar a la mitocondria a su degradación en un proceso denominado mitofagia (Figura 4, lado derecho, flechas rosas y moradas; Nguyen, Padman & Lazarou, 2016; Yamano, Matsuda & Tanaka, 2016).
Uno de los factores de transcripción clave en la respuesta mitocondrial a proteínas mal plegadas es ATFS-1 en Caenorhabditis elegans (Nargund, Pellegrino, Fiorese, Baker & Haynes, 2012) o ATF5 en mamíferos (Zhao et al., 2002). Este factor de transcripción tiene tanto una señal de localización mitocondrial como una de localización nuclear. En condiciones de homeostasis mitocondrial, ATFS-1 es importado hasta la matriz donde es degradado. Por el contrario, cuando hay un daño en la función mitocondrial que se refleja en la disminución del ΔΨ, ATFS-1 se acumula en el citosol y es translocado al núcleo para activar la transcripción de genes que permiten contrarrestar la disfunción en este organelo (Figura 4, lado derecho, flechas azules y rojas; Melber & Haynes, 2018; Qureshi, Haynes & Pellegrino, 2017). Entre los genes cuya expresión es inducida por ATFS-1 se encuentran chaperonas, proteasas y componentes de la maquinaria de importación mitocondrial (Lin et al., 2016; Nargund, Fiorese, Pellegrino, Deng & Haynes, 2015; Nargund et al., 2012).
Enfermedades asociadas a defectos de importación de proteínas mitocondriales
La mayor parte del conocimiento que tenemos sobre el funcionamiento y regulación de las maquinarias de importación de proteínas a la mitocondria proviene de estudios en organismos modelo. Sin embargo, se han identificado algunas enfermedades en humanos con mutaciones en componentes específicos de las maquinarias de importación, por lo que se ha planteado que los síntomas desarrollados son consecuencia de alteraciones durante la identificación y distribución de las proteínas mitocondriales. Hasta ahora, la mayoría de los reportes se limitan a describir la enfermedad y no ha sido posible comprender los detalles del mecanismo que subyace en el desarrollo de los síntomas observados. Es fundamental entender cómo las mutaciones actualmente identificadas afectan la importación de proteínas a la mitocondria, con el fin de proponer terapias para el tratamiento de estas patologías.
Enfermedades asociadas a la vía TOM-TIM23
Debido a que la mayor parte del proteoma mitocondrial reside en la membrana interna y en la matriz, muchas de las enfermedades mitocondriales descritas hasta este momento están asociadas con defectos en la importación de proteínas cuya translocación o inserción depende de TIM23 (Tabla I).
Tabla I Enfermedades en humanos que se han asociado con mutaciones en las maquinarias de importación de proteínas a la mitocondria.
La cardiomiopatía dilatada con ataxia DCMA (Dilated Cardio Miopathy with Ataxia, por sus siglas en inglés) es una enfermedad que está relacionada con mutaciones en el gen DNAJC19 cuyo correspondiente en S. cerevisiae es PAM18 (Al Teneiji, Siriwardena, George, Mital & Mercimek-Mahmutoglu, 2016; Davey et al., 2006; Ojala et al., 2012). Esta enfermedad es autosómica recesiva y se caracteriza por la presencia de cardiomiopatía y ataxia desde la niñez (la ataxia es la incapacidad de coordinar movimientos voluntarios asociada con daño neural). A nivel molecular, la enfermedad causa una disminución en el contenido de las proteínas mitocondriales y en la actividad de los complejos de la fosforilación oxidativa, presuntamente, como resultado de la disminución en la importación de proteínas a la mitocondria. De manera similar a DNAJC19, mutaciones en el gen MAGMAS, el ortólogo en levadura de PAM16, provocan la displasia SMDMDM (Spondylo Metaphyseal Dysplasia Megarbane-Dagher-Melki, por sus siglas en inglés; Mehawej et al., 2014; Short et al., 2012; Sinha, Joshi, Chiittoor, Samji & D'Silva, 2010). Esta enfermedad se caracteriza por distrofia muscular severa y defectos en el crecimiento y el desarrollo.
La encefalopatía epiléptica mitocondrial ha sido relacionada con mutaciones en la subunidad Tim50 del complejo TIM23 (Shahrour et al., 2017; Tort et al., 2019). Esta enfermedad está asociada con defectos en la función de la ATP sintasa, posiblemente derivado de la disminución en la función de TIM23 y en los defectos de importación de las subunidades que participan en la fosforilación oxidativa.
Otra enfermedad relacionada con la ruta TOM-TIM23 es la ataxia espinocerebelar recesiva autosómica, ocasionada por mutaciones en el gen PMPCA, que codifica para la subunidad a de la proteasa de la matriz MPP en humanos (Choquet et al., 2016; Jobling et al., 2015). Uno de los sustratos identificados con un procesamiento anormal resultado de estas mutaciones es la frataxina, proteína involucrada en la formación de centros hierro-azufre en la mitocondria y cuya alteración se asocia con la ataxia de Friedreich (Joshi et al., 2016). De manera similar, las mutaciones en el gen PMPCB, que codifica para la subunidad β de la MPP, se han relacionado con la neurodegeneración durante la infancia (Vögtle et al., 2018). Es probable que en estos casos exista un procesamiento deficiente de las MTS mitocondriales, que desencadena tanto una respuesta a proteínas mal plegadas, como la retención de los precursores mitocondriales en los complejos TOM y TIM23, comprometiendo así la importación de otros sustratos mitocondriales.
La vía de importación TOM-TIM23 también está afectada en neuropatologías. En la enfermedad de Parkinson se encontró que versiones de la α-sinucleína modificadas postraduccionalmente, inducen su interacción con el receptor Tom20 en las mitocondrias de las neuronas, tanto en modelos murinos como en muestras del tejido cerebral de pacientes (Di Maio et al., 2016); y esta asociación se correlaciona con una disminución en los complejos respiratorios, particularmente del complejo I (Devi, Raghavendran, Prabhu, Avadhani & Anandatheerthavarada, 2008; Franco-Iborra et al., 2018). Es probable que la asociación de la α-sinucleína con Tom20 comprometa su asociación con otros sustratos de importación. Estos hallazgos podrían explicar la disfunción mitocondrial previamente descrita en la enfermedad de Parkinson. En la enfermedad de Huntington se ha propuesto un modelo similar de patogénesis, donde la proteína huntingtina mutante es capaz de asociarse al complejo TIM23 e inhibir su función (Yablonska et al., 2019; Yano et al., 2014).
Enfermedades asociadas a la vía TOM-TIM22
El síndrome de distonía (contracción muscular involuntaria) y sordera humana DDS (human Deafness Distonia Syndrome, por sus siglas en inglés), está ligado al cromosoma X y es ocasionado por mutaciones en el gen DDP1, que codifica a una proteína equivalente a Tim8 de S. cerevisiae, una de las chaperonas del espacio intermembranal (Koehler et al., 1999; Tranebjaerg et al., 1995). Las mutaciones en DDP1 comprometen la función mitocondrial, posiblemente por el deterioro en la importación de proteínas a la mitocondria. El papel fundamental de las chaperonas del espacio intermembranal en las diversas rutas de importación podría explicar el fenotipo pleiotrópico encontrado en pacientes con este síndrome, que incluyen retraso mental, pérdida de la audición y ceguera (Jin et al., 1996).
El síndrome de Sengers es causado por mutaciones en el gen AGK, que codifica para la acil glicerol cinasa mitocondrial (Aldahmesh, Khan, Mohamed, Alghamdi & Alkuraya, 2012; Calvo et al., 2012; Mayr et al., 2012). Entre los síntomas de esta enfermedad se encuentran la cardiomiopatía hipertrófica, cataratas congénitas, acidosis láctica e intolerancia al ejercicio. Aunque inicialmente esta enzima se relacionó con la síntesis del ácido fosfatídico, recientemente se descubrió como una subunidad adicional del complejo TIM22 en humanos (Bektas et al., 2005; Kang et al., 2017; Vukotic et al., 2017). La observación de que existe una disminución de los transportadores de metabolitos en la membrana interna mitocondrial en pacientes con síndrome de Sengers, refuerza la idea de que esta enfermedad está relacionada con defectos en la importación mediada por el complejo TIM22 (Kang et al., 2017). A pesar de esto, aún no se ha dilucidado la relevancia del gen AGK en el desarrollo de la enfermedad de Sengers. Hasta este punto no se puede determinar si el rol de AGK ocurre sobre la biogénesis, la degradación, la estabilidad o la función de los transportadores de metabolitos en la membrana interna.
Enfermedades asociadas a la vía de MIA
Las mutaciones en el gen ALR, homólogo en S. cerevisiae del gen ERV1 , provocan un cuadro patológico que incluye miopatía y cataratas (Di Fonzo et al., 2009). Debido al papel de ALR/Erv1 en la vía de importación de proteínas al espacio intermembranal, es posible que la biogénesis de Mia40 esté afectada y, en consecuencia, también la importación de los sustratos que emplean esta vía (Ceh-Pavia, Ang, Spiller & Lu, 2014; Sztolsztener, Brewinska, Guiard & Chacinska, 2013).
Avances terapéuticos para el tratamiento de enfermedades mitocondriales
La terapia genética es una alternativa estudiada para curar enfermedades mitocondriales. En la actualidad, estas terapias se basan en el uso de vectores adeno-virales (VAVs) que permiten expresar un gen en un órgano blanco. Este tipo de terapias son más eficaces en las enfermedades causadas por la mutación de un solo gen, como es el caso de algunas ya mencionadas.
Los estudios pioneros en este campo demostraron la corrección de defectos musculares en ratones mutantes del gen ANT1 (transportador de la membrana interna mitocondrial), al expresar el gen silvestre mediante VAVs (Flierl, Chen, Coskun, Samulski & Wallace, 2005). De manera similar, se ha logrado expresar a la enzima sulfuro dioxigenasa (SDO) en el hígado de ratones knock-out de la SDO, lo que restableció la función hepática y la supervivencia de los animales (Di Meo et al., 2012). En otro ejemplo, el modelo murino knock-out del gen TYMP recrea el cuadro clínico observado en la encefalopatía mitocondrial neuro-gastrointestinal MNGIE (Mitochondial Neuro-GastroIntestinal Encephalopathy, por sus siglas en inglés). Para este último caso, también se han logrado revertir los cambios del metabolismo de la timidina con el uso de VAVs que permiten expresar al gen silvestre específicamente en el hígado (Torres-Torronteras et al., 2014; Torres-Torronteras et al., 2018). Por otro lado, se ha logrado revertir la degeneración de las neuronas del nervio óptico en modelos murinos Harlequin, mediante la administración vítrea de VAVs para expresar al gen silvestre AIF; y en modelos murinos con atrofia óptica dominante (Klein et al., 2002; Sarzi et al., 2018). Es notable que estas aproximaciones han escalado a estudios clínicos en humanos. Un estudio reciente en fase III, realizado en pacientes con distrofia retinal, demostró mejoría en la función visual después de la administración subretinal de VAVs permitiendo la expresión del gen silvestre RPE65 (Russell et al., 2017).
Estos estudios sustentan la viabilidad de expresar genes mitocondriales en pacientes que padecen enfermedades mitocondriales asociadas con la mutación de un gen nuclear en particular. El éxito de estas terapias dependerá principalmente del diseño del sistema de expresión utilizado, por un lado, para asegurar una expresión específica en el órgano blanco (serotipo viral y promotor utilizados) y, por el otro, para asegurar la integración del producto proteico en el subcompartimento mitocondrial deseado (adición de secuencias señal correctas tanto a nivel de proteína como de RNAm).
Mención aparte, son las enfermedades ocasionadas por mutaciones del genoma mitocondrial. Ya que, el genoma mitocondrial se resguarda en la matriz mitocondrial, su manipulación representa un reto de mayor complejidad. El genoma mitocondrial codifica proteínas altamente hidrofóbicas que son parte de los complejos respiratorios, por lo que su expresión desde el núcleo o "expresión alotópica", involucra el rediseño de la secuencia de aminoácidos de las proteínas para disminuir su hidrofobicidad y permitir su transporte y translocación exitosas a la mitocondria, pero conservando su asociación y función en los complejos respiratorios. Por todos estos aspectos, esta aproximación ha tenido resultados poco exitosos. Sin embargo, una estrategia diferente ha propuesto una solución a este problema con el uso de sistemas de edición genética con nucleasas híbridas denominadas TALENS (Transcription activator-like effector nucleases, por sus siglas en inglés). Las TALENS son nucleasas programables que cortan secuencias de DNA específicas, y han sido diseñadas para ingresar a la mitocondria y editar regiones del genoma mitocondrial, hasta el momento exitosas en modelos murinos de la mutación del RNAtAla. Estos estudios han demostrado que el genoma mitocondrial dañado deja de ser detectado después de la edición por las TALENS, sugiriendo una mejoría en la función mitocondrial. Sin embargo, es indispensable realizar más pruebas, en particular con la función de la OXPHOS, para asegurar que este proceso de edición genera los resultados esperados (Bacman et al., 2018; Gammage et al., 2018).
Conclusiones
Las mitocondrias son organelos que muy posiblemente se originaron a partir de un proceso endosimbionte entre una Archea y una α-proteobacteria, afectando radicalmente a ambos organismos. Una de las consecuencias más importantes fue la migración de los genes del genoma del huésped al genoma hospedero; por lo que se hizo imperativo el desarrollo de diferentes sistemas de transporte y distribución de las proteínas mitocondriales, actualmente codificadas mayoritariamente en el genoma nuclear. De esta forma, las células eucariontes establecieron diversas maquinarias para la translocación e inserción de las proteínas mitocondriales en los diferentes subcompartimentos de este organelo. Como cualquier proceso celular, el transporte y la importación de proteínas a la mitocondria puede alterarse por diferentes factores. Conocer las respuestas celulares que se desencadenan ha sido un tema de gran avance en los últimos años, teniendo como corolario general que hay cambios transcripcionales y traduccionales que conllevan a la síntesis de proteínas que ayudan a contrarrestar los efectos del daño. La mayoría de las mutaciones en las subunidades que componen las maquinarias de translocación son letales; sin embargo, algunas de estas mutaciones son viables, aunque generan fenotipos severos en las personas que las poseen, subrayando la importancia del correcto funcionamiento de las mitocondrias.
Declaración de contribución de los autores
Maria Clara Avendaño-Monsalve, José Carlos Ponce-Rojas y Soledad Funes planearon el artículo, revisaron el material bibliográfico, escribieron el texto y elaboraron las figuras.
Declaración de conflicto de interés
Los autores declaran no tener ningún conflicto de interés.

