











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El DNA se asocia con las proteínas histonas y con otras proteínas, conformando una estructura molecular conocida como cromatina. La unidad básica de la cromatina es el nucleosoma, que consiste de un fragmento del DNA asociado a un octámero de histonas, es decir, dos copias de cada una de las histonas H2A, H2B, H3 y H4. El nucleosoma humano está rodeado por 147 pares de bases de DNA, mientras que la histona H1 está situada fuera del octámero, promoviendo las interacciones entre los nucleosomas. La estructura primaria de la cromatina a menudo se denomina fibra de 11 nm o “collar de perlas” debido a la forma del nucleosoma y las secuencias espaciadoras entre ellos. Esta fibra a su vez forma la fibra de 30 nm o solenoide que son los nucleosomas empaquetados. Posteriormente, la forma de la cromatina puede adquirir estructuras de orden superior para compactar al genoma que incluyen: la fibra de 300 nm o dominios de bucles; la fibra de 700 nm que representa espirales condesadas de cromatina hasta el nivel más elevado de compactación que conforma a los cromosomas (Figura 1) (Lawrence, Daujat & Schneider, 2016).

El nivel de compactación de la cromatina es dinámicamente regulado, y constituye un punto de control para el acceso y el reclutamiento de factores para la replicación, reparación y transcripción del DNA, dentro del núcleo celular. A través de diferentes mecanismos que regulan la interacción entre las histonas y del DNA, se afecta el grado de compactación de la cromatina, conduciendo a una re-estructuración dinámica, que oscila entre un estado de compactación y descompactación.
Es importante considerar que el genoma humano (~3.2 x109 pares de bases) está empaquetado dentro del núcleo de las células, en gran medida por la conformación de la cromatina. En el genoma humano están contenidos los genes codificantes para proteínas (~2% del genoma, ~25,000 genes), además de secuencias que no codifican para proteínas (~98% del genoma). A partir de algunas de estas “secuencias no codificantes” se pueden generar transcritos conocidos como los RNA no codificantes (ncRNA, ~80% del genoma humano), que pueden actuar como factores reguladores de la expresión génica, de la traducción y del procesamiento del RNAm. El resto de las “secuencias no codificantes” corresponden a secuencias del DNA denominados elementos CIS, también implicados en la regulación de la expresión de los genes y en la estructuración del genoma (Elkon & Agami, 2017; Gomes et al., 2019; Moraes & Goes, 2016; Parasramka, Maji, Matsuda, Yan & Patel, 2016).
De manera interesante, todas las células que conforman un organismo contienen la misma información genética (genoma), pero presentan diferentes mecanismos moleculares de regulación de encendido y apagado de genes específicos.
Como resultado se establecen diferentes tipos de células, cada una con características morfológicas y funcionales particulares. De este modo, un hepatocito contiene la misma información genética que una neurona, o que cualquier otra célula del organismo, pero el patrón de genes que están encendidos y apagados en cada tipo celular es diferente, generando un perfil específico de expresión génica, de proteínas y de ncRNA propios para cada una de estas células. El encendido y apagado de genes es mediado por una interdependencia de diversos elementos, como: regiones regulatorias del DNA, la formación de complejos multiproteicos asociados al DNA, ncRNA y cambios en la conformación de la cromatina (Chen, Li, Subramaniam, Shyy & Chien, 2017; Pal & Tyler, 2016).
La compactación de la cromatina puede ser promovida o inhibida por modificaciones epigenéticas que son inducidas por diferentes factores biológicos y ambientales. Además, las modificaciones epigenéticas podrían influir en la personalidad y habilidades de los individuos (Tabla I). La epigenética (epi, del griego, que significa en/sobre), hace alusión a los cambios heredables en la expresión de los genes que no están asociados a la secuencia del DNA, sino a la conformación de la cromatina. Cada organismo tiene un epigenoma particular y a su vez cada grupo celular tiene su propio epigenoma que hace posible la activación y represión de genes específicos. Los ejemplos más visibles de la epigenética a nivel de un organismo se demuestran en gemelos monocigotos, que comparten la genética y sus rasgos fenotípicos, pero una epigenética distinta con diferentes personalidades, habilidades e incluso enfermedades. Al respecto, se han reportado diferencias en el perfil de metilación del DNA genómico, el patrón de acetilación de histonas y el transcriptoma de gemelos monocigotos durante su etapa adulta (Fraga et al., 2005). Los mecanismos epigenéticos permiten la adaptación al ambiente y son importantes en la salud y en procesos fisiológicos como el envejecimiento, pero también están implicados en el desarrollo de enfermedades, como las neurodegenerativas (Chen et al., 2017; Pal & Tyler, 2016; Yan, Matouk & Marsden, 2010).
Tabla I
| Comportamiento | Descripción | Modificación epigenética asociada | Referencia |
| Depresión | Ratones expuestos a un entorno de derrota social | Marcas de histonas represivas asociadas al gen BDNF | (Tsankova et al., 2006) |
| Esquizofrenia | Modelo de ratones reeler heterocigotos (HRM) | Alteraciones en los patrones de metilación del gen Reelina | (Tremolizzo et al., 2002) |
| Sociabilidad | Personas con baja metilación de oxitocina (OXT) tienen una mejor habilidad para reconocer expresiones faciales, que los que tienen más alta metilación de OXT, asociando las modificaciones epigenéticas de OXT con la sociabilidad humana. | Bajos niveles de metilación del gen OXT. | (Haas et al., 2016) |
| Victimización por intimidación y estrés en la infancia | Estudio longitudinal de gemelos monocigóticos. | El aumento de la metilación del DNA del gen transportador de serotonina (SERT). | (Ouellet-Morin et al., 2013) |
Generalidades del proceso de regulación de la expresión génica
La transcripción es el proceso en el que se producen transcritos del RNA a partir de una cadena molde del DNA y es el punto primario en el control de la expresión génica. La RNA polimerasa II (RNA pol II) es la responsable de generar transcritos a partir de genes específicos, que darán origen a las proteínas durante el proceso de traducción. Adicionalmente, la RNA pol II puede también generar RNAs que no codificarán para proteínas (ncRNA). En el proceso de transcripción se requieren los elementos CIS que están localizados dentro del genoma y contienen secuencias del DNA específicas. A su vez, los elementos TRANS se reclutan a las secuencias CIS para modular la transcripción. Los elementos TRANS no son secuencias del genoma, pero son productos de genes específicos, como factores de transcripción y co-reguladores transcripcionales. Los factores de transcripción (TF) son proteínas modulares que contienen un dominio de unión al DNA, caracterizado por la habilidad de reconocer y unirse a secuencias del DNA específicas a través de sus motivos estructurales, como los dedos de zinc, la estructura orquilla-beta (beta-hairpin), los homeodominios, así como estructuras del tipo: cierre de leucina, hélice-giro-hélice y hélice-bucle-hélice.
Los TF pueden clasificarse en específicos, generales y pioneros. Los TF específicos son activados por diferentes vías de señalización a través de modificaciones postraduccionales (como fosforilación), unión al ligando y/o oligomerización, que los conducen a un enriquecimiento dentro del núcleo para favorecer sus funciones. Algunos ejemplos de TF específicos son: Smad3, factor activado por fosforilación dependiente de la vía del TGF-beta (TGF-β); o el receptor de estrógenos alfa (ERα), activado por la unión de su ligando hormonal, el estradiol. Los TF generales o basales son proteínas conservadas, encargadas de interactuar con proteínas del complejo mediador (formado por alrededor de 26 subunidades en el humano) y primordialmente reclutar a la RNA pol II con el fin de conformar un complejo de pre-iniciación para la transcripción de un gen (Guertin & Lis, 2013; Guo, 2014; Lambert et al., 2018; Yin & Wang, 2014). En cambio, los TF conocidos como “pioneros” son proteínas que tienen la habilidad de unirse a sus sitios consenso del DNA, aun en la cromatina compactada por reconocer su topología (cuando otros TF no pueden hacerlo). Además, se ha sugerido que los TF pioneros pueden reconocer modificaciones al DNA y marcas de histonas asociadas con la apertura local de sitios de cromatina cerrados. De este modo, los TF pioneros tienen acceso al DNA antes del momento de la activación de la transcripción, y son importantes para iniciar el reclutamiento de la maquinaría de remodelación de la cromatina y los TF basales (Mayran et al., 2019; Zaret & Carroll, 2011).
En el proceso transcripcional, la RNA pol II requiere un sitio específico para iniciar la transcripción de un gen (TSS o transcription start site). Las secuencias del DNA denominadas promotor de un gen, comúnmente se localizan inmediatamente antes del TSS (río arriba del TSS). No obstante, algunos promotores abarcan el TSS, e incluso un par de bases río abajo del TSS. El promotor se puede dividir en un promotor proximal y un promotor basal, mínimo o core. Las secuencias del promotor proximal son reconocidas por TF específicos (activados por vías de señalización también específicas), para subsecuentemente promover la unión de los TF generales al promotor basal para la transcripción. El promotor basal, que incluye el TSS y se extiende ~35 nucleótidos más allá, río arriba y río abajo, puede tener secuencias consenso conservadas como la caja TATA, el elemento Iniciador, el elemento para el factor TFIIB, o regiones de islas CpG, a las que se unen los TF generales o basales (TFIIA, TFIIB, TFIID, TFIIE, TFIIF y TFIIH). La fosforilación de la RNA pol II por el TFIIH es requerida para iniciar la transcripción, y la subsecuente elongación y procesamiento del RNAm. El procesamiento incluye el splicing (remoción de intrones), la adición de la 7 metilguanosina trifosfato en el extremo 5´(CAP) y la poli-adenilación en el extremo 3´ (poli-A). Los RNAm procesados son transportados al citosol para ser traducidos en los ribosomas. Diversos mecanismos pueden regular la estabilidad o degradación de los RNAm, afectando la producción de proteínas, que además pueden ser modificadas postraduccionalmente para regular su abundancia y/o su función (Guertin & Lis, 2013; Roy & Singer, 2015).
Otras secuencias del DNA importantes son los enhancers, localizados a miles de bases de distancia, río arriba o abajo del TSS, y en cualquier orientación, dentro de regiones intra- o inter-génicas. Los enhancers reclutan diversos factores transcripcionales y coactivadores para incrementar la actividad transcripcional. Existen también regiones específicas en el genoma llamadas super-enhancers por contener múltiples enhancers asociados al control de la identidad celular. Un enhancer puede interactuar con múltiples promotores de manera específica mediante la formación de loops o bucles a través de las interacciones con el complejo multiproteico mediador, además de las proteínas cohesinas que estabilizan la interacción enhancer-promotor. Adicionalmente, las secuencias insulator son elementos del DNA, que actúan como secuencias barrera, impidiendo la expansión de marcas de heterocromatina. Los insulators también interrumpen la comunicación entre secuencias regulatorias enhancers y sus promotores. CTCF (por su nombre en inglés CCCTC- binding factor) es una proteína multifuncional de expresión ubicua conformada por 11 dedos de zinc, y es considerada una “proteína insulator” por su capacidad de unión a secuencias insulator, participando en la regulación transcripcional. CTCF también interactúa con otras proteínas como las cohesinas y forma asas de cromatina o loops que afectan la organización del genoma. Adicionalmente CTCF puede también actuar como un represor transcripcional. (Erokhin, Vassetzky, Georgiev, & Chetverina, 2015; Kagey et al., 2010; Kim et al., 2015; Rao et al., 2014). Un parálogo de CTCF es BORIS (por su nombre en inglés Brother of the regulator of the imprinted site), que tiene una expresión enriquecida en los espermatocitos y en algunos tipos de cáncer como el de mama, de próstata, de colon y endometrial (Martin-Kleiner, 2012).
Los co-reguladores o co-factores transcripcionales, son proteínas que no poseen la capacidad de asociarse directamente al DNA, pero son reclutados a las regiones regulatorias a través de su asociación con los TF específicos. De manera importante, los co-reguladores pueden tener la habilidad de modificar la estructura de la cromatina o de reclutar proteínas con esta actividad. Así, los co-reguladores son clave en los mecanismos de regulación transcripcional y epigenética. Los co-reguladores pueden ser co-activadores que inducen modificaciones en la cromatina para hacerla más abierta y disponible para los procesos transcripcionales. En contraste, los co-represores median modificaciones en la cromatina asociadas con su compactación para evitar la expresión de los genes (Dasgupta, Lonard & O’Malley, 2014; Reiter, Wienerroither & Stark, 2017) (Figura 2).

Elaboración personal.
Figura 2 Elementos CIS y TRANS en la regulación de la expresión génica. A) Los factores transcripcionales (TF) son elementos TRANS reclutados a elementos CIS del DNA como promotores (distal y mínimo/basal o core) y enhancers. B) El complejo mediador actúa como puente molecular entre los complejos multiproteicos regulatorios asociados a la región promotora y enhancer. C) Los co-reguladores se asocian al DNA a través de los TF, y tienen habilidad de asociarse con otros correguladores y modular la estructura de la cromatina. Los co-reguladores pueden ser co-activadores y co-represores, que inducen a una cromatina permisiva o no permisiva a la transcripción, respectivamente.
Fundamentos de la Epigenética
La cromatina menos condensada o abierta se denomina eucromatina y se caracteriza por contener genes transcripcionalmente activos. En contraste, la cromatina más condensada o compacta es inaccesible para la transcripción y se conoce como heterocromatina. La heterocromatina se compone principalmente de secuencias repetidas como el DNA satélite (secuencias altamente repetidas en tándem), SINES (por sus siglas en inglés: short interspersed nuclear elements) de entre 100-500 pb de longitud, y LINES (por sus siglas en inglés: long interspersed nuclear elements) de entre 6,000-7,000 pb de longitud. Se denomina heterocromatina facultativa a las regiones de eucromatina que llegan a silenciarse; y heterocromatina constitutiva a las regiones que se mantienen silenciadas y que están enriquecidas en los telómeros y centrómeros. Así, la heterocromatina constitutiva establece regiones estructurales asociadas con la estabilidad del genoma, mientras que la heterocromatina facultativa puede reactivarse como eucromatina. De manera interesante, la eucromatina está distribuida principalmente en la región central del núcleo, mientras que la heterocromatina se encuentra hacia la periferia nuclear y alrededor del nucléolo (Gonzalez-Sandoval & Gasser, 2016).
Algunas regiones de la cromatina pueden mantener una interacción con otras regiones distales de la misma cromatina, o con estructuras subcelulares como la lámina nuclear. De este modo, se han identificado diferentes unidades de cromatina que están en una proximidad espacial a las que se denominan TADs (por su nombre en inglés topologically associated domains). También, se han determinado dominios que se conocen como LADs (por su nombre en inglés laminaassociated domains) por la asociación de la cromatina con la lámina nuclear. Tanto los TADs como los LADs regulan la organización de la cromatina dentro del núcleo, afectando la regulación transcripcional. Es importante resaltar, como previamente se mencionó, que el DNA al estructurarse en cromatina tiene diferentes grados de compactación, siendo el más alto nivel el que conduce a la formación de los cromosomas mitóticos. Además, durante la interfase (cuando el DNA no está condensado en cromosomas mitóticos), los cromosomas se distribuyen en los denominados territorios cromosómicos de una manera que no parece ser azarosa dentro del núcleo celular (Gonzalez-Sandoval & Gasser, 2016) (Figura 3).

Elaboración personal.
Figura 3 TADs, LADs y territorios cromosómicos. Los cromosomas están distribuidos en regiones específicas dentro del núcleo de las células, en los llamados territorios cromosómicos. Los dominios de la cromatina denominados LADs se asocian a la lámina nuclear. La interacción espacial entre dominios de cromatina distales son denominados TADs. Cromatina inactiva transcripcionalmente en TADs están distribuidas hacia la periferia nuclear (compartimento B) en contraste con las regiones de cromatina activa en TADs (compartimento A). Proteínas estructurales como CTCF y complejos de cohesinas son importantes en la estabilización de loops de la cromatina locales.
La regulación epigenética se refiere a los mecanismos que modulan la estructuración de la cromatina, para generar permisividad o no permisividad al DNA, regulando así la expresión génica. Estos mecanismos incluyen: las modificaciones postraduccionales de las histonas, las modificaciones al DNA y la remodelación de la cromatina (Cremer & Cremer, 2010; Chen et al., 2017) (Figura 4). En general, la eucromatina se caracteriza por presentar una mayor acetilación en las histonas así como bajos niveles de metilación del DNA; mientras que la heterocromatina se caracteriza por contener altos niveles de metilación del DNA (Richards & Elgin, 2002). Específicamente, la heterocromatina constitutiva se caracteriza por presentar la marca de histonas H3K9me3, H3K27me1, H4K20me2/3, H3K64me3, mientras que las marcas de histonas H3K27me3 y H2AK119Ub se han vinculado a la heterocromatina facultativa (Saksouk, Simboeck & Dejardin, 2015).

Elaboración personal.
Figura 4 Modificaciones epigenéticas. La compactación y descompactación de la cromatina es dinámica conduciendo al apagado y prendido de la expresión génica, respectivamente. La re-estructuración de la cromatina es modulada por modificaciones de las histonas como la acetilación asociada a una cromatina abierta, y la desacetilación de histonas asociada con la compactación de la cromatina y represión transcripcional. La metilación del DNA se caracteriza por la unión de grupos metilos a citosinas (5mC), y está vinculada con una cromatina cerrada, mientras que una desmetilación activa conlleva a la formación de un intermediario de 5-hidroximetilcitosina (5hmC) que se vincula a una cromatina menos compactada, asociación de co-activadores y actividad transcripcional.
Los remodeladores de la cromatina son proteínas encargadas de reestructurar los nucleosomas. Esta reestructuración o remodelación se refiere al desplazamiento de los nucleosomas de una manera dependiente del ATP, generando su reposicionamiento y creando, como consecuencia, zonas del DNA libres de nucleosomas, con un mayor acceso para los complejos multiproteicos reguladores. No obstante, otras zonas del DNA pueden ser protegidas por los nucleosomas como resultado del desplazamiento inhibiendo el acceso a la maquinaria transcripcional y evitando su expresión (Langst & Manelyte, 2015). Los remodeladores de la cromatina de la familia de SWI/SNF son complejos multiproteicos que han sido ampliamente estudiados, dividiéndose a su vez en diferentes complejos de acuerdo a las subunidades que los conforman (Mashtalir et al., 2018).
La conformación de la cromatina de un organismo puede ser retenida por una “memoria celular” que es establecida por el grupo de genes Polycomb (PcG) y Trithorax (TrxG) durante el desarrollo de un organismo. Los complejos PcG y TrxG están conformados por diferentes co-reguladores y ejercen funciones antagónicas. El complejo PcG establece modificaciones de la cromatina para el silenciamiento de la expresión génica, mientras que el complejo TrxG establece marcas de activación en enhancers y promotores, las cuales son leídas por complejos modificadores de la cromatina asociados con la activación de la expresión génica (Tie et al., 2014).
En esta revisión nos enfocamos en la modificación de las histonas y del DNA como mecanismos asociados con el envejecimiento del cerebro.
Modificación de histonas. Como se mencionó, las histonas son proteínas básicas que conforman a los nucleosomas. El extremo N-terminal de las histonas (15-30 aa) es poco estructurado, flexible y ubicado fuera del centro del nucleosoma, por lo que se les conoce como colas de las histonas, susceptibles a diversas modificaciones como la acetilación, la metilación, la fosforilación o la ubiquitinación, entre otras. Al respecto, los procesos de acetilación y desacetilación de las histonas han tenido mayor atención. La acetilación es catalizada por enzimas como las acetiltransferasas de las histonas (HAT), mientras que la desacetilación es dirigida por las desacetilasas de las histonas (HDAC). Estos procesos son reversibles y ocurren en residuos de lisina específicos. La unión de un grupo acetilo a la lisina de una histona neutraliza su carga positiva, disminuyendo así la afinidad por el DNA, y relajando la estructura de la cromatina para promover la expresión génica. En contraste, la remoción del grupo acetilo por las HDACs incrementa la carga positiva de las histonas y con ello la afinidad por el DNA, favoreciendo una estructura más compactada de la cromatina asociada con la represión en la expresión de los genes. Además de la acetilación/ desacetilación de histonas, los residuos de lisina (K), arginina (R) e histidina (H) de las colas de las histonas pueden ser modificados por metilación (Susztak, 2014).
Otras modificaciones como la fosforilación y la ubiquitinación, entre otras, han sido menos estudiadas; pero son importantes para regular el reclutamiento de complejos proteicos a la cromatina. Se ha comprobado la existencia de combinaciones específicas de ciertas modificaciones que conforman un código de histonas que puede ser crítico en la dinámica estructural de la cromatina y por consiguiente en la expresión génica (Susztak, 2014). Un ejemplo es la serotonilación de histonas que se refiere a la unión covalente de la serotonina a la glutamina 5 de la histona 3 (H3Q5). Esta modificación ocurre en los nucleosomas con la marca H3K4me3 mediante la enzima transglutaminasa 2 (TGM 2), generando la combinación H3K4me3Q5ser, asociada con una expresión génica activa en células neuronales al reclutar el factor TFIID (por su nombre en inglés TATA-box-binding protein) sobre los promotores (Farrelly et al., 2019). Otro ejemplo, se presenta en la transcripción del gen FOSL1, por un mecanismo que involucra una serie de modificaciones de histonas consecutivas y cooperativas. La histona 3 acetilada, en la zona del enhancer FOSL1, es fosforilada por PIM1 en respuesta al suero, generando la marca H3S10ph. Esta marca es reconocida por la proteína 14-3-3, la cual recluta a la acetiltransferasa MOF, generando la marca H4K16ac. Como resultado se genera el código H3K9acS10ph/H4K16ac, importante para la elongación del transcrito del gen FOSL1 (Zippo et al., 2009).
Cabe mencionar que existen también variantes de las histonas, que pueden reemplazar a las histonas “canónicas” asociadas al nucleosoma, afectando la estructura y estabilidad de éste, la dinámica de la cromatina y la expresión génica (Lardenoije et al., 2015; Lawrence et al., 2016). Algunos moduladores epigenéticos (readers) contienen dominios estructurales que les confiere la habilidad de reconocer las modificaciones postraduccionales sobre las histonas y modificaciones en el DNA, realizadas por enzimas específicas (writers), por ejemplo, las proteínas que contienen bromodominios leen histonas acetiladas y las proteínas que contienen cromodominios leen lisinas metiladas. Además, algunas proteínas (erasers) son capaces de remover las marcas realizadas por las proteínas “writers”, formando parte de una dinámica de modificaciones reversibles. De este modo diversas proteínas son reclutadas a la cromatina formando complejos multiproteicos que mantienen los circuitos requeridos para el encendido o apagado de los genes (Yun, Wu, Workman & Li, 2011).
Modificaciones al DNA. En la metilación del DNA, los residuos de citosina del dinucleótido CpG (5 ‘C-fosfato-G-3’) se modifican por la adición de un grupo metilo. Muchos genes presentan regiones ricas de este dinucleótido denominadas islas CpG. La metilación del DNA es una modificación química que es llevada a cabo por el DNA metiltransferasas (DNMT), que incluyen a DNMT1, DNMT2, DNMT3a, y DNMT3b. La presencia de grupos metilo en el DNA puede afectar la unión de los factores de transcripción a sus secuencias consenso en regiones regulatorias. Además el DNA metilado puede ser reconocido por proteínas MBP (por su nombre en inglés Methyl-CpG-binding proteins), las cuales comúnmente reclutan correpresores y HDACs para promover la compactación de la cromatina (Kemme, Marquez, Luu & Iwahara, 2017; Mehler, 2008).
La desmetilación activa del DNA es principalmente mediada por enzimas TET (Ten-Eleven Translocation of 2-oxoglutarate and iron-dependent dioxygenase), las cuales oxidan a la 5 metilcitosina (5mC) a la 5 hidroximetilcitosina (5hmC), 5 formilcitosina y la 5 carboxilcitosina, seguido por la escisión de bases mediada por TDG (thymine DNA glycosylase) y reparación del DNA para generar una citosina no modificada. En otras palabras, en la hidroximetilación del DNA, un átomo de hidrógeno del grupo metilo de la 5mC es reemplazado por un grupo hidroxilo que genera un hidroximetilo. El descubrimiento de 5hmC y las enzimas TET sugieren importantes mecanismos de regulación en los distintos procesos biológicos, incluyendo aquellos que controlan la función del sistema nervioso, debido a que, en los mamíferos, los niveles de 5hmC son más altos en el cerebro. Por lo tanto, la hidroximetilación del DNA es considerada un paso intermedio de la vía de desmetilación del DNA, aunque también se considera como una señal para factores moduladores de la cromatina. De esta manera, mientras que la metilación del DNA es generalmente asociada con la represión transcripcional, el DNA con marcas de 5hmC está relacionado con activación de la transcripción, y se ha reportado enriquecido en elementos enhancer activos (Kaas et al., 2013; Kriaucionis & Heintz, 2009; Michaeli et al., 2013; Tahiliani et al., 2009).
La epigenética y el envejecimiento del cerebro
El envejecimiento es un proceso natural ocurre en todos los seres vivos, e implica cambios graduales en todo el organismo, incluido el nivel molecular. En los humanos, es claramente visible cómo el envejecimiento está asociado con disfunciones en diversos procesos cognitivos como pérdida de la memoria, de la atención, y alteraciones en las funciones de ejecución. Estas funciones cognitivas son reguladas por el hipocampo y la corteza prefrontal (CPF), al parecer afectadas durante el envejecimiento (Barter & Foster, 2018). El cerebro de los mamíferos contiene más de un billón de neuronas, que están asociadas con las células gliales (astrocitos, oligodendrocitos y microglia). Las neuronas y las células gliales están interconectadas, y durante el envejecimiento suelen ocurrir alteraciones en estas células, que dañan las conexiones sinápticas. Los sellos o “hallmarks” del envejecimiento incluyen una serie de cambios a nivel celular y molecular: 1) la disfunción mitocondrial, y la acumulación intracelular de moléculas afectadas por oxidación; 2) la desregulación del metabolismo energético, autofagia y degradación vía proteosoma; 3) alteraciones en los mecanismos reparadores del DNA; 4) daño en la neuroplasticidad y la actividad en las redes neuronales; 5) desregulación del calcio neuronal; y 6) inflamación. Además, la senescencia y el acortamiento de los telómeros, se consideran dos marcadores de envejecimiento (Horvath & Raj, 2018).
Senescencia
Los mecanismos que causan el envejecimiento no se conocen con exactitud, pero uno que ha sido propuesto es el establecimiento de la senescencia celular, como un proceso que conduce al arresto del ciclo celular (Campisi, 2013; Martinez-Cue & Rueda, 2020; Rizos, Haferkamp & Scurr, 2017; Scurr, Haferkamp & Rizos, 2017). La senescencia es un estado no proliferativo, pero viable y metabólicamente activo de las células, y es acompañada por una actividad secretora pro-inflamatoria. La senescencia actúa como un mecanismo de defensa de las células, evitando la proliferación de células transformadas o con daño en su DNA. Sin embargo, la acumulación de células senescentes parece contribuir al envejecimiento, al afectar a las células no senescentes, alterando la capacidad regenerativa y funcional de los tejidos. Las células al adquirir un fenotipo secretor asociado a la senescencia (SASP), pueden liberar diversos factores que estimulan la inflamación crónica, y contribuir a la degeneración relacionada con la edad (Kritsilis et al., 2018; Martinez-Zamudio, Robinson, Roux & Bischof, 2017). De manera importante, en la senescencia celular se ha observado la formación de dominios heterocromáticos facultativos, denominados focos heterocromáticos asociados a la senescencia (SAHF) y a la disminución en la expresión de los genes que codifican para las histonas. A pesar de ello, las células senescentes pueden también presentar una disminución en el número de marcas de heterocromatina represivas como la metilación del DNA y la metilación de las histonas, incluidas las marcas H3K9me3, H3K27me3 y H4K20me3 (Nacarelli, Liu & Zhang, 2017; Sidler, Kovalchuk & Kovalchuk, 2017). Esto implica cambios drásticos en el patrón del encendido y apagado de genes específicos durante la senescencia celular asociada al envejecimiento.
Durante la senescencia las células adquieren una forma plana e irregular, con alteraciones funcionales en el rearreglo del citoesqueleto, retículo endoplásmico y mitocondrias (Kritsilis et al., 2018). En la senescencia también se presenta una alteración de la proteostasis (homeostasis de proteínas), produciendo alteraciones en: la síntesis, plegamiento, degradación y agregación de proteínas (Ishikawa & Ishikawa, 2020; Martinez-Cue & Rueda, 2020). Los marcadores moleculares de la senescencia incluyen: SA-β-gal lisosomal, acumulación de lipofuscina en citoplasma, inhibidores de cinasas dependientes de ciclinas como p15INK4B, p16INK4A, p19 INK4D y p21WAF1/Cip1, así como un fenotipo inflamatorio (Bussian et al., 2018; Campisi, 2013; Hunter, Arendt, & Brayne, 2013; Ishikawa & Ishikawa, 2020; Kritsilis et al., 2018; Masaldan, Belaidi, Ayton & Bush, 2019; Medeiros & LaFerla, 2013).
Además, a nivel genético, cuando el individuo envejece, los telómeros se acortan, como resultado de cada división celular, lo que puede llevar a una inestabilidad cromosómica (Baker & Petersen, 2018; Calcinotto et al., 2019; Campisi, 2013; Rizos et al., 2017; Scurr et al., 2017). La senescencia celular se asocia con el acortamiento de los telómeros, la activación de los oncogenes, y daño al DNA.
De manera interesante, el envejecimiento del humano contrasta con otros organismos que no envejecen, como la rata topo desnuda (Heterocephalus glaber), la cual se caracteriza por poseer una longevidad de hasta 30 años, y una alta resistencia al cáncer comparado con ratas y ratones. Las hembras no tienen disminución en la fertilidad, ni riesgo de mortalidad asociado a la edad (ley de Gompertz). Por lo tanto, se considera a la rata topo como un mamífero que no envejece (Buffenstein, 2008; Delaney, Kinsel & Treuting, 2016a; Delaney et al., 2016b; Ruby, Smith & Buffenstein, 2018). La actividad reproductiva y fisiológica de este animal se mantiene el 80% de su vida, y solo los animales mayores a 28 años presentan pérdida muscular, y acumulación de lipofuscina (Edrey, Hanes, Pinto, Mele & Buffenstein, 2011). Este modelo animal tiene una alta cantidad de peroxidación lipídica, proteínas carboniladas y daño oxidante al DNA (Perez et al., 2009), y una expresión de genes involucrados en la reparación del DNA más elevada que en los ratones (MacRae et al., 2015). Además, las ratas topo tienen un alto nivel de expresión del gen que codifica a la subunidad C del complejo succinato deshidrogenasa (Sdhc), siendo una proteína asociada a la membrana mitocondrial que forma parte de uno de los complejos enzimáticos de la cadena respiratoria (Yu et al., 2011). Dado que los niveles de metilación del genoma se han asociado con la edad de un organismo (ver relojes epigenéticos, más adelante), se ha propuesto evaluar la acumulación del DNA metilado en sitios específicos del genoma de la rata topo desnuda para entender su envejecimiento (Lowe et al., 2020).
Epigenética de un envejecimiento patológico y no patológico
La pérdida progresiva de la estructura y función neuronal en el envejecimiento favorece la neurodegeneración, relacionándolas con enfermedades como Alzheimer, Parkinson y Huntington. Estas enfermedades se caracterizan por la generación de proteínas mal plegadas y agregación de péptidos (beta-amiloide/tau, alfa-sinucleína y huntingtina, respectivamente), alteraciones metabólicas y de vías de señalización, así como neuroinflamación (Kritsilis et al., 2018).
Una conformación predominante de eucromatina (cromatina abierta) y una mayor actividad transcripcional se han vinculado con la marca H4K16ac. Un marcado enriquecimiento de esta modificación se detecta durante el envejecimiento sano de los lóbulos temporales del cerebro humano. Contrariamente, la marca H4K16ac se reduce en el envejecimiento con la neurodegeneración y aparición de la Enfermedad de Alzheimer. Por lo tanto, una mayor predisposición a enfermedades neurodegenerativas en el envejecimiento, podría asociarse a la disminución de la marca H4K16ac (Nativio et al., 2018).
Similarmente, otra proteína que parece ser importante en el envejecimiento es el factor de transcripción REST (factor de transcripción de silenciamiento RE1 o NRSF). Clásicamente, REST recluta co-represores que compactan la cromatina y como resultado generan un silenciamiento de la transcripción de genes, entre ellos genes que promueven la muerte de las células neuronales. REST también puede estar asociado con la activación transcripcional, debido a cambios en la cromatina, promoviendo la expresión de los genes de protección a las neuronas contra el estrés oxidante (Christopher, Kyle & Katz, 2017; Hwang & Zukin, 2018). REST se expresa en neuronas corticales y del hipocampo en el envejecimiento sano de los humanos. En contraste, la expresión del gen REST disminuye durante la neurodegeneración. Además, la detección de REST se reduce en el núcleo celular, mientras que aparece con proteínas mal plegadas patológicas en autofagosomas. Los datos sugieren que REST podría tener una actividad clave neuroprotectora durante el envejecimiento del cerebro. De este modo, REST podría vincularse con la preservación y longevidad, mientras que su disminución parece ser un factor de riesgo crítico para la progresión de enfermedades neurodegenerativas (Lu et al., 2014). Adicionalmente, REST es producido en neuronas senescentes y se ha propuesto como un marcador de senescencia neuronal in vitro (Piechota et al., 2016).
Por otra parte, se ha observado, principalmente en el hipocampo y en la corteza frontal, una disminución de la expresión de diferentes enzimas como DNMT1, DNMT3a1 y DNMT3a2 así como de TET1 y TET2 durante el envejecimiento. En cambio, la proteína MBP2 está incrementada en muestras postmortem en el cerebro de personas envejecidas. Estos resultados indican que durante el avance de la edad ocurren cambios en la expresión de los mediadores epigenéticos asociados con el perfil de metilación/desmetilación del DNA en el sistema nervioso central (Pagiatakis, Musolino, Gornati, Bernardini & Papait, 2019; Torres, Kouro & Kerr, 2019).
Asimismo, los niveles de metilación del genoma se han asociado con la edad, debido a que el perfil global de metilación de islas CpG se modifica con el envejecimiento. Esta estimación de la edad con base en los niveles de metilación del genoma se conoce como “reloj epigenético”, considerado un proceso innato e inevitable de las células en el envejecimiento (Horvath & Raj, 2018; Zhang, Qu, Liu & Belmonte, 2020). Los relojes epigenéticos se basan en algoritmos matemáticos que correlacionan el estatus de metilación del DNA de células, tejidos u órganos específicos con la edad cronológica del individuo. Por ejemplo, el reloj epigenético propuesto por Horvath se basa en el perfil de metilación de islas CpG de varias fuentes de células, tejidos y órganos. Se ha propuesto, que particularmente el patrón de metilación del DNA de las células de la sangre se asemeja al del cerebro durante el envejecimiento (Horvath et al., 2012). Un perfil similar de metilación del DNA en la sangre, del riñón y del músculo esquelético se observó también en el cerebro (Day et al., 2013). Además, el análisis del DNA metilado en 387 regiones del cerebro humano en edades de 1 a 102 años mostraron que la metilación de los genes PIPOX, DPP8, RHBDD1, FLJ21839, y PTGER3 está altamente asociada con la edad cronológica (Hernandez et al., 2011).
El perfil de metilación del DNA de la corteza humana se relaciona con la neurodegeneración ligada al envejecimiento. Una “edad epigenética acelerada” es producto de una carga amiloide, la presencia de placas difusas y un declive en la memoria (Levine, Lu, Bennett & Horvath, 2015). Por lo tanto, en algunos casos puede existir una edad “epigenética acelerada”, que consiste en una mayor edad epigenética que la edad cronológica. De manera interesante, en una “parabiosis heterocrónica” (PH), los sistemas circulatorios de animales jóvenes y maduros se unen quirúrgicamente, lo que facilita el intercambio de factores secretados y células inmunológicas de la sangre. La PH puede atenuar los daños asociados con la edad en varios tejidos, incluyendo el cerebro. Por lo tanto, es probable que la restauración de factores solubles de organismos jóvenes sea capaz de reajustar el reloj epigenético de organismos envejecidos (Horvath & Raj, 2018; Zhang et al., 2020).
Cabe resaltar que en las enfermedades neurodegenerativas, como la Enfermedad de Alzheimer, existe una desregulación en el estatus de la expresión de las enzimas y marcas de metilación del DNA, así como en la modificación de histonas en comparación con un envejecimiento no patológico (Cadena-del-Castillo et al., 2014; Fransquet & Ryan, 2018; Nativio et al., 2018; Zhou & Xu, 2015). Si bien es cierto que estas enfermedades se han asociado con una edad epigenética acelerada, también se ha reportado una asociación con la hipometilación de algunas regiones regulatorias de genes específicos. Por ejemplo, las regiones regulatorias de genes como DSCAML1, BACE1, PSEN1 y APP están hipometiladas en neuronas de pacientes con Enfermedad de Alzheimer (en comparación con neuronas de un envejecimiento no patológico), promoviendo la expresión de estos genes y la progresión de la patología (Li et al., 2019; Xiao, Liu & Jiao, 2020). Por consiguiente, el análisis del epigenoma del cerebro durante una vejez sana, podría contribuir a prevenir alteraciones que conduzcan a una vejez con neurodegeneración ligada a patologías.
Conclusiones
De manera general, la regulación transcripcional consiste en el reclutamiento y ensamblado de complejos multiproteicos específicos que se unen a regiones regulatorias del DNA para favorecer o inhibir la expresión de los genes. Dentro de estos complejos existen co-reguladores que tienen la habilidad catalítica o de asociación con las enzimas para modificar la cromatina. Los cambios en la estructura de la cromatina vía su compactación y des-compactación, tienen impacto directo sobre la represión y la actividad transcripcional, respectivamente. La regulación en la expresión de los genes determina la morfología y la función celular, así como la respuesta celular ante diferentes factores, por lo que es de gran importancia no sólo en la fisiología de la célula, sino también en los procesos vinculados a una patología.
Durante el envejecimiento, existe una acumulación de células senescentes que pueden llegar a contribuir a la neurodegeneración. Además, durante el envejecimiento, tanto en células senescentes como no senescentes, ocurren cambios epigenéticos, que podrían determinar una vejez sana de nuestro cerebro o una vejez cerebral vulnerable a la enfermedad (Figura 5). Un reloj epigenético acelerado puede ser un factor de riesgo para desarrollar enfermedades neurodegenerativas, por lo que su entendimiento podría elucidar nuevas estrategias sobre los mecanismos involucrados en estas neuropatologías.
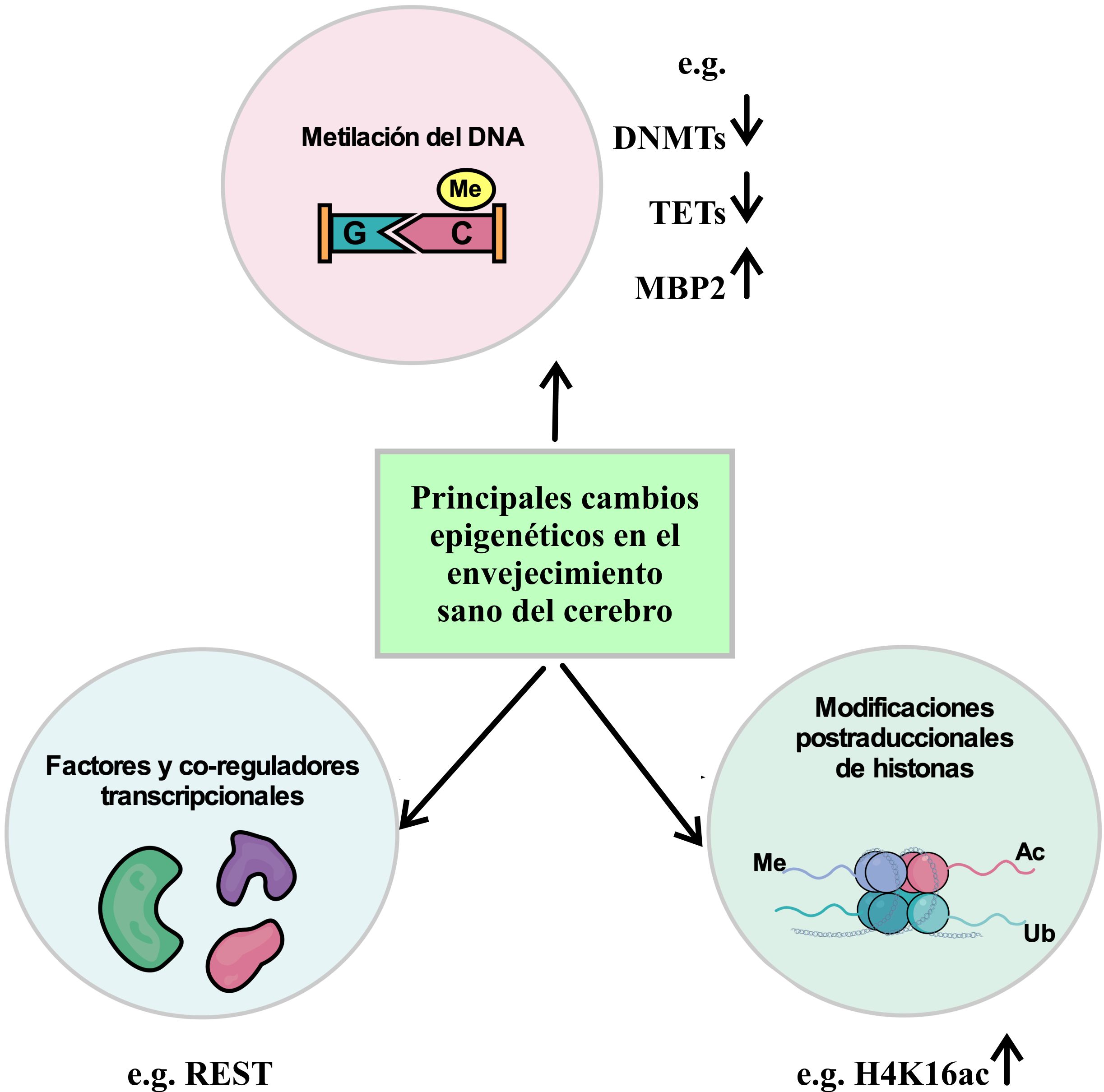
Elaboración personal.
Figuras 5 Principales modificaciones epigenéticas en el envejecimiento sano del cerebro humano. Además de la cooperación de factores de transcripción y co-reguladores en la modulación de la expresión génica durante el envejecimiento, cambios en diferentes modificaciones epigenéticas, como el estatus de metilación del DNA, y modificaciones de las histonas, tienen un efecto sobre la dinámica de la cromatina.
Hasta ahora la detección de la marca H4K16ac, y la expresión y abundancia de REST parecen generar neuroprotección cerebral en la vejez, mientras que su reducción se muestra como un factor de riesgo para el desarrollo de enfermedades neurodegenerativas. Se requiere de un mayor conocimiento sobre los cambios epigenéticos que contribuyen a un envejecimiento saludable, así como mayores estudios sobre las modificaciones de la cromatina que están asociadas a la degeneración de las células del sistema nervioso central.

