











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introducción
La geometría molecular es la disposición tridimensional de los átomos. Al trabajar con una molécula el principal objetivo es encontrar la conformación de menor energía debido a que las estructuras moleculares en su conformación ideal determinan la función y reacción de la mismas [17,29].
Algunos de los métodos experimentales usados para determinar una estructura proteica son: la cristalografía de rayos X y la espectroscopia por RNM, presentando la problemática con los errores sistemáticos.
Otra forma de determinar la estructura es prediciendo los datos a través de métodos computacionales y modelos de interacciones moleculares. Estos métodos se basan en la información de la secuencia de aminoácidos que se encuentra recopilada en un banco de proteínas por sus siglas en inglés (PDB) [5,25], a partir de esta información se predice la estructura tridimensional teniendo en cuenta que la estructura terciaria nativa de una proteína corresponde a la conformación de mínima energía del sistema.
Las moléculas de proteínas y polipéptidos, están formados por cientos y cientos de átomos, y como la superficie de energía potencial depende de las coordenadas atómicas, el número de variables necesarias para el problema es 3N-6 (donde N es el número de átomos en una molécula), cuando aumenta el número de átomos, crece exponencialmente el número de mínimos locales [21], por lo que este problema es considerado como un problema NP-completo, lo que significa que no existe algoritmo conocido que lo resuelva en tiempo polinomial [11].
Debido a que la función de energía potencial es multimodal, no convexa, y con un gran número de variables hace que este problema continúe siendo de interés para las áreas de química, física y cómputo y un tema de investigación extremadamente activo ya que cada una de ellas considera el problema desde distintos puntos de vista [7].
Dada la complejidad del problema, muchos investigadores dedicados a resolver problemas de optimización, han estado desarrollado diversos algoritmos con diferentes técnicas, dentro de las que presentaron mejores resultados con un tiempo computacional aceptable son las metaheurísticas [12] [27], tales como los algoritmos genéticos [14,15], recocido simulado [1], búsqueda tabú [13], GRASP [10] y colonia de hormigas [18] entre otras.
Sin embargo en la última década, los algoritmos más populares han sido los algoritmos evolutivos [31], ya que por el éxito de sus soluciones se consideran una poderosa herramienta para atacar los problemas de optimización en general.
Los algoritmos evolutivos (AEs) [4,26], utilizan mecanismos inspirados en la evolución natural, como la reproducción, recombinación, mutación y selección para trabajar con una población de individuos que representan soluciones candidatas para el problema.
La población es sometida a ciertas transformaciones y posteriormente a un proceso de selección el cual favorece a los mejores individuos. Cada ciclo de transformación y selección constituye una generación, de tal forma que después de un determinado número de generaciones se espera que el mejor individuo de la población se encuentre lo más cercano al óptimo global.
Como los algoritmos evolutivos están enfocados en la optimización global y su éxito depende del equilibrio entre la exploración y explotación del espacio de búsqueda [8], es recomendable incorporar un algoritmo de optimización local para evitar quedarse atrapado en un óptimo local.
En particular en este trabajo se propone utilizar un AE para dar solución al problema de optimización geométrica de una molécula de proteína.
Este algoritmo parte de una estructura inicial obtenida experimentalmente y es caracterizada mediante un modelo simplificado que considera únicamente los ángulos diedros φ y ψ que forman la cadena principal [2,6], de esta manera es posible manipular la geometría y evaluarla por medio de una gráfica Ramachandran [19,23], en la que se distinguen diversas regiones válidas para la función de optimización.
2. Estructura molecular
Las proteínas son moléculas muy complejas formadas por un conjunto de aminoácidos, unidos mediante un enlace peptídico.
Los aminoácidos son los elementos constitutivos de las proteínas y su estructura general consta de un grupo carboxílico y un grupo amino unidos al mismo átomo de carbono-α por medio de un enlace peptídico, así, todos los aminoácidos estándar que contienen las proteínas son α-aminoácidos como se puede observar en la Fig. 1.
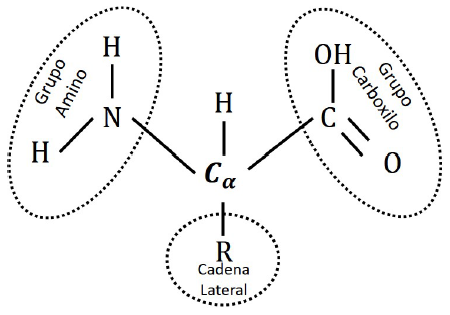
Al carbono α se le unen otros dos sustituyentes: un átomo de hidrogeno y una cadena lateral (o grupo R) que es lo que hace diferente a cada aminoácido y por medio de la cual se le asigna nombre y una clasificación.
Cuando se unen dos aminoácidos por medio de un enlace peptídico se les llama dipeptido, el enlace peptídico es la eliminación de una molécula de agua entre el ácido carboxílico y el grupo amino y a la porción de cada aminoácido que permanece en la cadena se denomina residuo de aminoácido.
En la estructura de un péptido se diferencian las cadenas laterales de la cadena principal (o esqueleto peptídico), ya que están formadas por los átomos de los enlaces peptídicos de cada residuo de aminoácido. El grupo amino N-terminal y el carboxilato C-terminal son parte también de la cadena principal.
Las cadenas que contienen unos pocos residuos de aminoácidos se denominan oligopéptidos y si la cadena es más larga (>~15-20 residuos), se llama polipéptido. Los polipéptidos mayores de ~50 residuos se denominan proteínas.
La organización de una proteína viene definida por cuatro niveles estructurales denominados: estructura primaria, secundaria, terciaria y cuaternaria.
La estructura primaria es la secuencia lineal de aminoácidos que integran una proteína, en ésta se indica la cantidad y el tipo de los aminoácidos que la forman así como el orden en el que se encuentran unidos; la estructura secundaria se refiere a la conformación local de ciertas regiones de un polipéptido, es decir a los patrones de plegamiento regulares que adopta la cadena polopeptídica, las estructuras secundarias estables que se distribuyen en la proteína son: hélices α y láminas β; la estructura terciaria, es la disposición espacial de todos los átomos y está determinada por los plegamientos que tiene la cadena proteica por encima de la estructura secundaria que determina la forma tridimensional de la proteína.
Las cadenas polipeptídicas no solo son lineales sino que también están dobladas en formas compactas que contienen espirales, regiones en zigzag, giros y asas.
Su forma tridimensional o conformación es un ordenamiento espacial de átomos que depende de la rotación de uno o varios enlaces. La conformación de una molécula como la de una proteína, puede cambiar sin que los enlaces covalentes se rompan.
Como cada residuo de aminoácido tiene múltiples conformaciones posibles y considerando que hay numerosos residuos en una molécula de proteína, por tanto, cada molécula ofrece una cantidad astronómica de configuraciones.
Bajo condiciones fisiológicas, cada proteína se dobla en una forma estable llamada conformación nativa y la función biológica depende por completo de esta estructura [16].
2.1. Modelo simplificado de un polipéptido
Una de las principales complicaciones para las técnicas de minimización energética molecular es el tamaño del espacio de búsqueda, sin embargo se han propuesto métodos eficientes mediante el uso de aproximaciones continuas en modelos de estructuras proteicas que presentan buenos resultados.
Cada residuo en la estructura secundaria es caracterizado por sus ángulos diedros de la cadena principal y una de las 20 posibles cadenas laterales de los aminoácidos unidos a un átomo Cα.
La estructura tridimensional está determinada por las coordenadas moleculares internas que consisten en longitudes de enlaces 'l' (definidas por cada par de átomos), ángulos de enlace θ (definida por la unión de cada 3 átomos en la cadena principal) y finalmente por los ángulos diedros (ϕ, ψ y ω) donde ϕ da la posición de C relativa a los tres átomos previos en la cadena principal los cuales son C-N-Cα, ψ da la posición de N relativa a los tres previos átomos de la cadena principal N-Cα-C y ω da la posición de de Cα relativa a los primeros tres átomos previos de la cadena principal, tal como se ilustra en la Fig. 2.

Algunos parámetros no cambian de forma independiente pero si pueden considerarse como fijos ya que se pueden mover sobre un rango de valores muy pequeño y por lo tanto el objetivo es ajustarlos a sus valores ideales, por ejemplo ω se ajusta a la conformación trans ω = 180° de esta manera solo nos quedaremos con n-1 pares de ángulos diedros (ϕ, ψ) de la cadena principal para la representación del modelo reducido, además los átomos que van de Cα al siguiente Cα pueden ser agrupados en planos peptídicos rígidos y por tanto no se requieren parámetros adicionales para expresar la posición tridimensional [9].
2.2. Geometría molecular
La geometría o estructura de una proteína es la disposición tridimensional de los átomos que constituyen la molécula. Es importante porque determina muchas de sus propiedades y se puede calcular por procedimientos cuánticos o por métodos semiempíricos de modelado molecular, las moléculas grandes tienen múltiples conformaciones estables que difieren en su geometría.
La posición de cada átomo se determina por la naturaleza de los enlaces químicos con los que se conecta a los átomos vecinos. La geometría molecular puede describirse por las posiciones de estos átomos en el espacio, considerando la longitud de enlace de dos átomos conectados, el ángulo de enlace de tres átomos conectados y el ángulo diedro de tres enlaces consecutivos.
El proceso de minimización de energía es muy costoso computacionalmente debido a la cantidad de operaciones que se tienen que realizar en cada evaluación a la geometría de la molécula.
2.2.1. Longitud de enlace
Dadas las posiciones de los átomos en un espacio tridimensional están dadas por las coordenadas (x,y,z) se realiza el cálculo para la distancia de la siguiente manera:
Sea el átomo A tiene las coordenadas
Lo mismo para el cálculo de la norma:
2.2.2. Ángulo de enlace
El producto interno de dos vectores normalizados u y v, es el coseno del ángulo entre los vectores, esto es:
Considerando dos átomos A y B con coordenadas
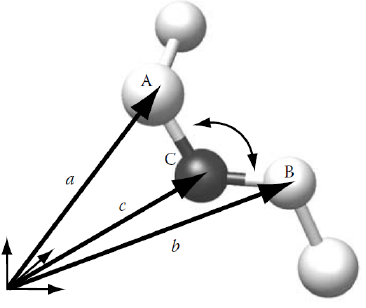
2.2.3. Ángulo Diedro
Al considerar los átomos de la cadena principal:
Esto no sucede con los ángulos diedros ya que estos se deben a una acción giratoria alrededor de un solo enlace y son los principales responsables de definir la posición de los átomos de la cadena principal en el espacio tridimensional, sin embargo hay que considerar que los cambios en los ángulos diedros no afectan los valores de los ángulos de enlace entre los átomos sucesivos de la cadena principal.
Un ángulo diedro esta definido por una secuencia de cuatro átomos enlazados consecutivamente, como se puede observar en la Fig. 4a.

Fig. 4 Ángulo diedro Phi (a) Representación de la disposición tridimensional de los átomos y la formación del ángulo alrededor del enlace N - Cα y (b) El ángulo φ esta formado por la intersección de la normal del plano Ci+1, Ni, Cαi y la normal del plano Ni, Cαi Ci
En general se puede observar la secuencia de los átomos que definen los dos planos que se intersectan en una línea que coincide con el enlace N - Cα y al ángulo que se forma entre esos dos planos es llamado ángulo φ(phi).
Análogamente en la Fig. 5a, se puede apreciar que en el enlace Cα - C que forma el ángulo ψ(psi), hay otro ángulo diedro que se debe considerar.

Fig. 5 Ángulo diedro Psi (a) Representación de la disposición tridimensional de los átomos y la formación del ángulo alrededor del enlace Cα-C y (b) El ángulo ψ formado por la intersección de la normal del plano Ni, Cαi Ci y la normal al plano Cαi, Ci, Ni+1
Este ángulo se forma por los átomos Cαi Ci Ni Ci-1, el cual tiene como eje de giro el enlace C - N y es llamado ω(omega), su enlace peptídico tiene un carácter parcial de doble enlace y por lo general tiene una configuración trans o ω = 180°.
En las Figs. 4b y 5b, se ilustra el ángulo diedro que es el ángulo formado por la intersección de los planos p1 y p2 que se encuentra a partir de los vectores normales a los planos.
Por ejemplo, para los vectores
De la misma forma para el plano p2 con
divididos ambos por su norma
Con respecto al signo, por convención se supone que un ángulo diedro está en el rango [ -π, π], sin embargo, el cálculo de la función nos conduce a un ángulo en el rango [ 0, π ], por tanto, es necesario ajustarlo.
Para el ajuste se considera el vector normal del plano definido por los primeros tres átomos y se calcula el producto interno con el vector que va del tercer átomo al último, si el producto interno es positivo, entonces el signo del diedro es positivo, en otro caso es negativo.
3. Transformación de coordenadas
Aplicaciones como la alineación estructural o el plegamiento de proteínas, requieren modificar la conformación de una proteína girando parte de la misma alrededor de algún eje arbitrario, esto es posible aplicando transformaciones como la rotación o la traslación de las coordenadas espaciales de sus átomos.
Dado un punto p que se encuentra en el plano xy, al
rotar p un ángulo θ, la posición final estará dada por el vector
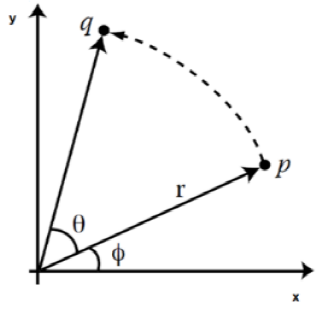
Sin embargo la rotación de los átomos en una molécula, requiere de una rotación sobre un eje arbitrario como se puede apreciar en la Fig. 7.

3.1. Rotación de un punto alrededor de un eje arbitrario
Dado un eje arbitrario definido por un vector unitario
donde el cosθ está representado por cθ, el senoθ por sθ y 1-cosθ por Fcθ.
4. Algoritmo evolutivo
Los algoritmos evolutivos (AEs) son una herramienta poderosa utilizada para encontrar soluciones muy cercanas al (óptimo global en problemas complejos con un espacio de búsqueda muy grande en un tiempo computacional razonable.
Los AEs, se basan en las reglas de la naturaleza, donde los individuos más aptos de la población sobreviven y son elegidos para reproducir la siguiente generación, los descendientes heredan características de los padres.
En estos algoritmos también existe la posibilidad de que individuos con poca aptitud sean elegidos para reproducirse y que las mutaciones nos proporcionen diversidad en la población.
Cada algoritmo enfatiza características diferentes para llevar el proceso de evolución de forma exitosa, algunos se concentran en la mutación como el principal operador, mientras que otros en la recombinación o la selección, sin embargo el comportamiento del algoritmo está determinado por la relación de explotación y exploración que se mantienen a lo largo de la ejecución.
Debido a que en la mayoría de los casos es difícil predecir si los individuos caerán en las zonas de interés, en este trabajo se propone dirigir la búsqueda a esas zonas mediante una técnica local de agrupamiento.
4.1. Representación de los individuos y función de aptitud
En este trabajo los individuos representa el conjunto de ángulos diedros (φ,ψ,ω) que componen la cadena principal de la molécula como se puede observar en la Fig. 8 y están codificados con números reales que van de (-π, π) para moverse dentro de los valores permitidos de acuerdo a la gráfica de Ramachandran.

La función de aptitud evalúa la energía de los individuos y los individuos más aptos son aquellos que presentan la menor energía.
Por tanto, es necesario considerar que la estructura de una molécula puede caracterizarse por las posiciones de sus átomos. Para una estructura y un estado electrónico dado, una molécula tiene una energía específica.
Para calcular la energía potencial del sistema mediante el método de mecánica molecular, en su forma más general este método considera los campos de fuerza siguientes:
donde Etotal es la energía total del sistema, (Er ) es la energía asociada al estiramiento de los enlaces, (Eθ) la energía de la curvatura de los ángulos de enlace, (EΦ) la energía de los ángulos de torsión y Enb a la energía de las interacciones no enlazadas, en algunas ocasiones se requiere agregar ciertos términos especiales que no abordaremos en este trabajo.
En su mayoría las ecuaciones de mecánica molecular son similares en cuanto a los términos:
donde
El potencial de Lennard-Jones 6-12 es el más usado para las interacciones de Van der Waals (EvdW) en (Enb), tal que:
como las moléculas bioquímicas están a menudo cargadas se agrega un término de energía electrostática (Eelec) a la (Enb):
donde Aij y Bij son parámetros de van der Waals y D es una constante dieléctrica molecular que explica la atenuación ambiental de la interacción electrostática entre los dos átomos con la carga puntual qiqj.
De acuerdo al estado del arte los métodos de mecánica molecular presentan una filosofía en común pero difieren en el número y tipo de las funciones de energía potencial que componen el campo de fuerzas. Los campos de fuerza más comunes son MM2/3 [3], AMBER [30] o CHARMM [20].
En este trabajo el principal cambio de energía proviene de los potenciales angulares dado que las distancias entre los átomos permanecen sin cambio. Así la función de aptitud es:
4.2. Representaciones de los ángulos diedros en la gráfica de Ramachandran
Una vez caracterizada la cadena principal por los ángulos diedros phi φ y psi ψ, se podrá cambiar la conformación de la molécula mediante el giro en torno a los enlaces sencillos de los Cα.
El ángulo φ (phi) es la rotación en torno al enlace Cα - N y el ángulo ψ (psi) es la rotación en torno al enlace C - Cα. Como estos son los únicos grados de libertad que presenta la estructura, la conformación de la cadena polipeptídica queda completamente definida cuando se conocen todos los valores de phi φ y de psi ψ.
Ramachandran y colaboradores [22], dedujeron que las cadenas peptídicas principales se encuentran en ciertas regiones del espacio de configuración de los ángulos diedros (φ, ψ) y esto permite la evaluación de la naturaleza geométrica de las estructuras proteicas.
Las combinaciones de φ y ψ en un residuo pueden representarse en una Gráfica de Ramachandran como la de la Fig. 9, esta gráfica está dividida en zonas prohibidas y permitidas, no todos los pares de (φ, ψ) están permitidos, ya que muchas rotaciones de estos enlaces implican interacciones estéricas desfavorables entre átomos del mismo residuo u otros próximos [24].

Las zonas doblemente sombreadas representan las conformaciones (combinaciones de φ y ψ) para las que no existe ningún impedimento estérico. Las conformaciones con sombreado sencillo, corresponden a las conformaciones para las que hay algún impedimento, pero que pueden presentarse si la distorsión se compensa mediante interacciones en otra parte de la proteína.
La Tabla 1, muestra como los valores de los ángulos dependen la forma de la cadena lateral de cada uno de los aminoácidos.
Tabla 1 Parámetros geométricos estructurales regulares
| Estructura Secundaria | Ángulos de torsión | ||
|---|---|---|---|
| φ | ψ | ω | |
| Hélice α
dextrogira |
-57 | -47 | -47 |
| Hoja β
paralela |
-119 | +113 | +113 |
| Hoja β antiparalela |
-139 | +135 | +135 |
Para formar distintas estructuras se pueden aplicar rotaciones a los planos e ir midiendo los cambios de energía en el sistema de tal forma que se puedan observar cuantitativamente las preferencias conformacionales.
4.3. Implementación
En la Fig. 10, se muestran los pasos que sigue la estrategia evolutiva propuesta para la optimización geométrica. Esta estrategia esta dividida en tres etapas principales las cuales se describen a continuación.
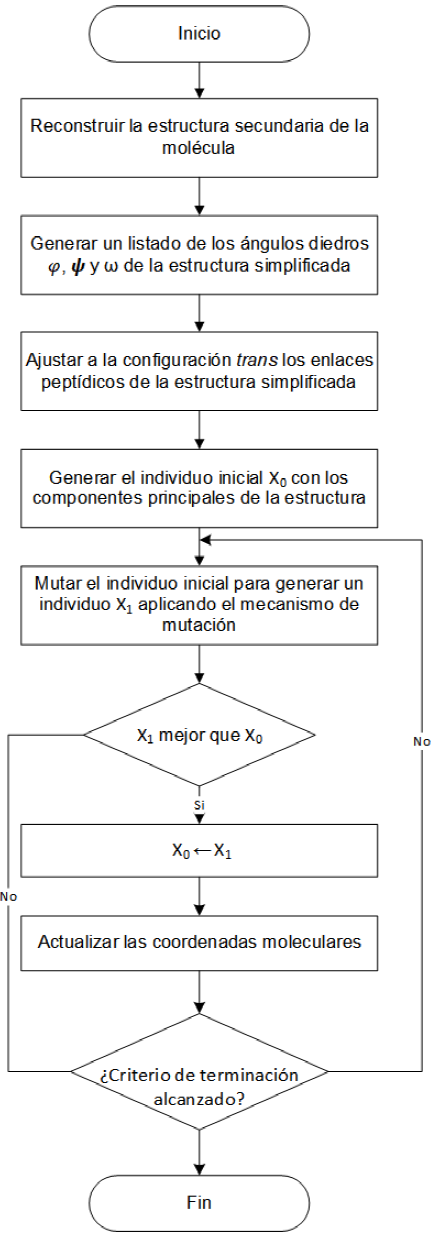
Etapa 1: Reconstrucción del modelo simplificado
○ Se reconstruye la estructura secundaria de una molécula de proteína (2CDS), a partir de las coordenadas moleculares proporcionadas por la base de datos (PDB), las cuales son obtenidas mediantes cristalografía de rayos X o resonancia magnética.
○ Una vez reconstruida la estructura de la molécula se identifican los ángulos diedros φ, ψ, y ω que componen la cadena principal y se genera un listado, ya que son los componentes principales con los que trabaja la estrategia evolutiva y el mecanimo de mutación.
○ Como se índice) en la sección 4.2, para modificar la conformación de la molécula se tiene que aplicar rotaciones en torno a los enlaces relacionados con Cα y para ellos es necesario ajustar todos los planos peptídicos a la configuración trans tal que ω = 180.
Etapa 2: Estrategia evolutiva
○ La estrategia evolutiva opera construyendo un individuo padre x0 como se mencionó en el apartado 4.1.
○ Con el individuo padre se genera un descendiente x1 mutándolo de acuerdo al operador de mutación como se muestra en la Fig. 11.
○ Ambos individuos x0 y x1 se evalúan mediante la función de aptitud correspondiente a la ecuación 11.
○ Se compara la aptitud de ambos individuos y se queda como padre para la siguiente generación el mejor de los individuos y se deshecha el otro.
○ En el caso de que el hijo reemplace al padre es necesario actualizar las coordenadas moleculares para que la geometría de la molécula corresponda a los cambios provocados por las mutaciones.
○ Estos pasos se repiten de forma iterativa y el algoritmo finaliza una vez que permanezca sin cambios por un número predeterminado de generaciones.
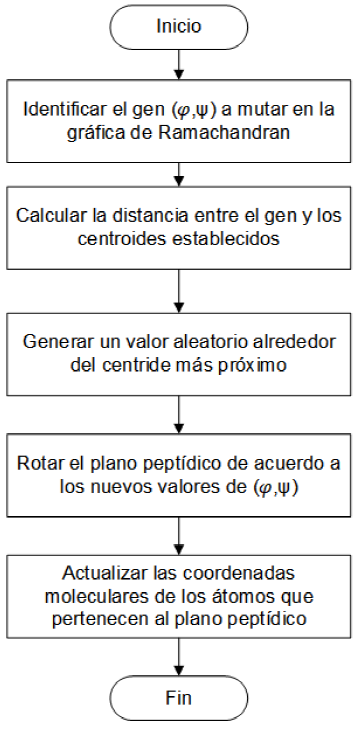
Etapa 3: Operador de mutación
El operador de mutación trabaja sobre un algoritmo de agrupamiento y la gráfica de Ramachandran descrita en el apartado 4.2, de tal manera que el nuevo individuo que representa una posible solución se mueva sobre las regiones más favorables dentro del espacio de búsqueda y que se controle la exploración y explotación del espacio de búsqueda.
El algoritmo de agrupamiento propuesto para este trabajo se basa en el algoritmo de agrupamiento K-Means [28], sin embargo, los centroides propuestos son fijos y están definidos de acuerdo a las regiones favorables para los ángulos diedros que describen al individuo.
5. Resultados
En esta sección presentamos el resultado de la optimización geométrica para la molécula 2CDS, obtenida del PDB. Esta molécula cuenta con 1001 átomos, los cuales fueron utilizados para la reconstrucción del modelo simplificado, al inicio la molécula presento una energía de 160.86 Kcal/mol, valor que se tomó como referencia para el individuo de la primera generación y de ahí en adelante el algoritmo busco mejorarla en cada iteración hasta converger en una energía de 136.02 Kcal/mol.
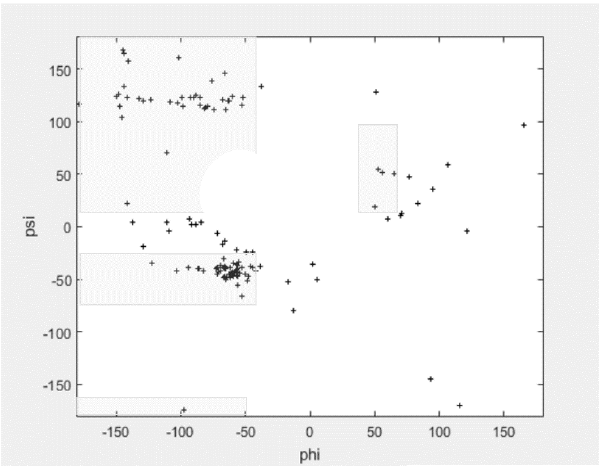
En la Fig. 12, se muestra la representación de los genes que corresponden al individuo inicial como puntos (φ,ψ), en la dispersión de los datos se puede apreciar que los puntos que se ubican en la región sombreada son los que se encuentran dentro de los valores permitidos.

Posteriormente se realizaron una serie de pruebas considerando modificar solo uno de los ángulos para ver el comportamiento del algoritmo.
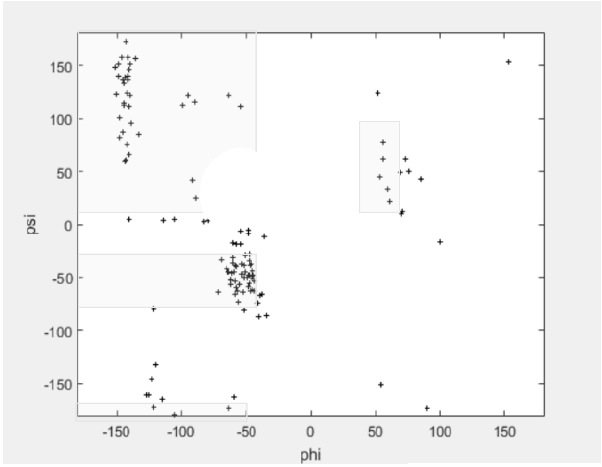
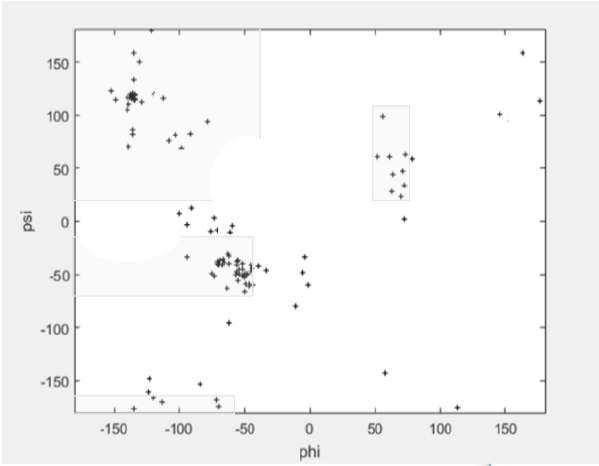
En la Fig. 13, se muestra una dispersión en forma horizontal, en la Fig. 17 de forma vertical y en la Fig. 18 se elige de forma aleatoria, presentando mejores resultados.
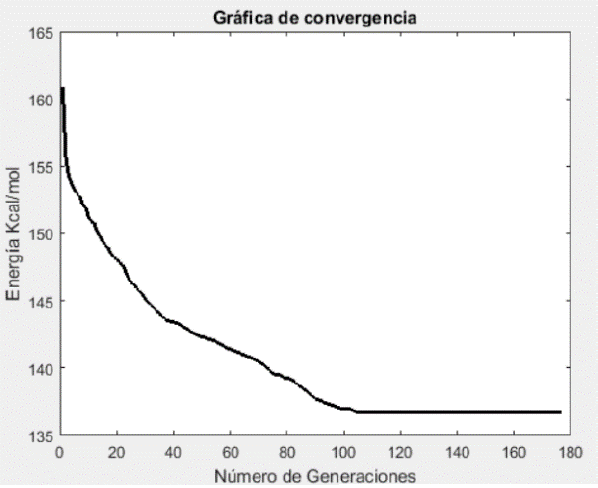
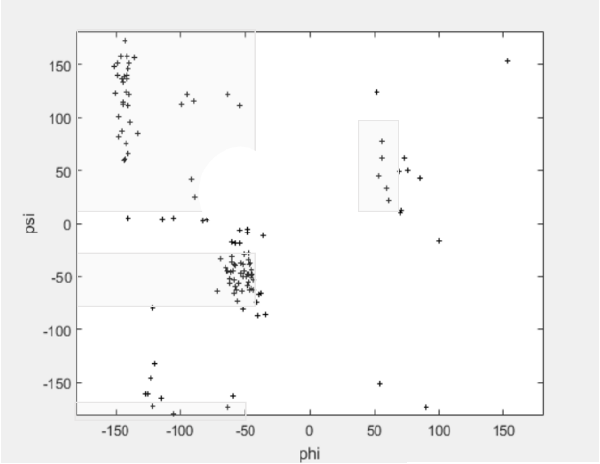

En la gráfica de convergencia representada por la Fig. 19, se puede observar como el algoritmo mejora la estructura geométrica de la molécula en cada iteración, de tal forma que disminuye la energía y en el momento que se mantiene estable el algoritmo converge.

6. Conclusiones
El manejo de la estructura secundaria por medio de un modelo molecular simplificado que toma los ángulos diedros como componentes principales, ayudo a reducir de forma significativa el espacio de búsqueda, de tal forma que de trabajar la optimización en un espacio multidimensional y con una gran cantidad de variable, la optimización se trabajó en un espacio bidimensional con 3n variables, donde n es el número de aminoácidos que componen la molécula de la proteína, sin embargo a pesar de esto el número de posibles configuraciones seguía siendo muy elevado por lo que la combinación del algoritmo evolutivo con la técnica de agrupamiento permitió mantener un equilibrio entre la exploración y la explotación y finalmente dirigir la búsqueda únicamente sobre las regiones donde se ubican los valores más favorables para encontrar las estructuras que están más cercanas al óptimo.

