











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Pholisma culiacanum es una holoparásita de raíz, con importancia agronómica, alimenticia, económica, cultural, biológica y evolutiva y representa un modelo adecuado para conocer los cambios en el proceso de la fotosíntesis del autotrofismo al parasitismo (Bungard, 2004).
Las plantas parásitas representan 1 % de las angiospermas, de ellas hay unas cuatro mil especies, agrupadas en 265 géneros en el mundo y 36 de estos se localizan en México. En Sinaloa se encuentran seis géneros de plantas parásitas, entre ellos Pholisma, que se distribuye desde el sur de Culiacán hasta los estados de California y Arizona en EE.UU. De las tres especies del género, P. culiacanum es endémica de los estados de Sonora y Sinaloa, donde el área circundante a Culiacán es el posible centro de origen de la especie y del género (Yatskievych et al., 1986).
El objetivo de esta investigación fue determinar la variabilidad genética en poblaciones de P. culiacanum, con secuencias genéticas de la región intergénica (TrnL-F) de ADN de cloroplasto, e identificar zonas con potencial para explotación in-situ y conservación de esos recursos genéticos.
Materiales y métodos
Muestreo
Para localizar las poblaciones de P. culiacanum se consultaron bases de datos: GIFB (Global Information of Biodiversity, 2013), Missouri Botanical Garden, Universidad de Arizona, Escuela de Biología y Facultad de Agronomía de la Universidad Autónoma de Sinaloa, Sonora (ERNO-UNAM) y se exploraron áreas que cumplen con las características de distribución. En cada población se recolectaron de 10 a 30 individuos (separados por un mínimo de 10 m), se geo referenciaron y almacenaron a -80 °C hasta realizar la purificación del ADN.
Amplificación
Para la amplificación se evaluaron los métodos de extracción de ADN propuestos por Doyle (1991), Porebski et al. (1997), Sá et al. (2011), Cota-Sánchez et al. (2006), Krizman (2006), Sánchez-Hernández (2006) y Amani et al. (2011). La metodología adecuada se basó en la descrita por Azmat et al. (2012).
Al carecer de información de la variabilidad genética de P. culiacanum, evaluamos varias regiones de ADN del cloroplasto reportadas en otras parásitas, en las que se detectó la variabilidad suficiente para realizar estudios de genética de poblaciones (Taberlet et al., 1991, dePamphilis et al., 1997; Zuber et al., 2000; Heinze, 2007; Amico et al., 2009; Nickrent et al., 2009). Con base en los resultados, los iniciadores que seleccionamos fueron c-Tab F y f-Tab R de Taberlet et al. (1991), esos amplifican la región intergénica del Trn L-F. El protocolo de amplificación usado fue por PCR tipo touch-down (Amico et al., 2007), con una modificación en las temperaturas de alineamiento de 48 a 50 °C y de 52 a 54 °C. Las reacciones de amplificación se realizaron en un termociclador Techne Unit Genius® y se visualizaron en geles de agarosa al 1 %, teñidos con bromuro de etidio (0.003 %), en un transluminador de luz UV.
Los productos de amplificación se secuenciaron en el HighThroughput Sequencing Solutions del Departamento de Ciencias Genómicas de la Universidad de Washington en Seattle, Washington, EE.UU. Para la edición y el alineamiento de las secuencias se utilizaron los programas CodonCode Aligner (CodonCode Corporation, 2009), Finch TV 1.4.0 (Geospiza Inc., 2004-2006) y el ClustalW multiple aligment del paquete informático BioEdit (Hall, 1999).
Análisis de la variabilidad
Los programas DnaSP (Librado y Rosas, 2010) y ARLEQUIN 3.1 (Excoffier, 2005) se utilizaron para calcular los índices de diversidad genética: 1) diversidad nucleotídica “π” (Nei, 1987, Hedrick, 2005, Hartl y Clark, 2007); 2) diversidad haplotípica Hd considerando y sin considerar indels como sitios informativos (Nei, 1987); 3) componentes de la variación debida a diferencias entre y dentro de las poblaciones (AMOVA). Para evaluar si las poblaciones se ajustan al modelo de aislamiento por distancia se realizaron análisis de correlación y de regresión lineal entre las distancias genéticas y geográficas con el modelo de Maximum Composite Likelihood sin considerar indels en MEGA 4.0, Tamura et al. (2007) y Microsoft 2007.
Red de haplotipos
Las secuencias de haplotipos se generaron con el programa DnaSP sin considerar los sitios alineados con gaps, datos perdidos o sitios invariables (monomórficos). El programa Network 4.6.1.1 (Fluxus Technology, 2004-2012) se utilizó para construir redes de haplotipos (Bandelt et al., 1999; Polzin et al., 2003) con secuencias de Lennoa como grupo raíz.
Filogenia de haplotipos
El árbol filogenético se generó en MEGA 4.0 (Tamura et al., 2007) con el método Neighbor-Joining, con un boostrap de mil replicas. Las distancias genéticas se calcularon con el método de Maximum Composite Likelihood sin considerar indels. Secuencias de Lennoa sp. se utilizaron como grupo externo.
Resultados y discusión
Muestreo
Nuestra exploración del área de distribución de P. culiacanum mostró siete poblaciones: cinco en Sinaloa y dos en Sonora. De los individuos recolectados, de cada población se seleccionaron 10, para realizar la purificación del ADN. En algunos lugares de distribución reportados (GIF, 2013) no se localizaron poblaciones debido al cambio de uso de suelo, de bosque primario a zonas agrícolas o de explotación forestal (Ranchería Los Álamos, Cerro siete gotas en Culiacán, San Blas), al desarrollo de vías de comunicación (Carretera al sur de Culiacán, Carretera Los Mochis-Obregón, Carretera Altata-Dautillos), al desarrollo turístico-inmobiliario (zona costera de Nuevo Altata) y a la consecuente fragmentación de los ecosistemas.
Amplificación
De todas las muestras secuenciadas, 70 pertenecían a P.culiacanum y dos a Lennoa madreporoides. El tamaño del fragmento amplificado fue de 735 nucleótidos, 261 presentaron gaps o fueron datos perdidos, 151 fueron sitios mono mórficos y 323 polimórficos, 251 de ellos presentaron dos tipos de sustituciones, 69 tres tipos y tres de ellos cuatro tipos. La longitud del producto de amplificación, las regiones no amplificadas (rps432-trnL [UAA], trnT [GGU]-trnE [UUC], atpB-rbcL) y las amplificadas con niveles bajos de variación (trnT [UGU] - trnL [UAA], rpl32F - trnL [UAG] y trnT [UGU] - trnL [UAA]) sugieren que en P. culiacanum, como en otras parásitas, existe reducción del tamaño del cloroplasto comparado con el tamaño original de la planta del tabaco (1560 nucleótidos) (Bungard, 2004).
Análisis de la variabilidad
Las siete poblaciones evaluadas son polimórficas, con 11 haplotipos diferentes (dos a cuatro por población) sin considerar indels como sitios informativos (Figura 1).
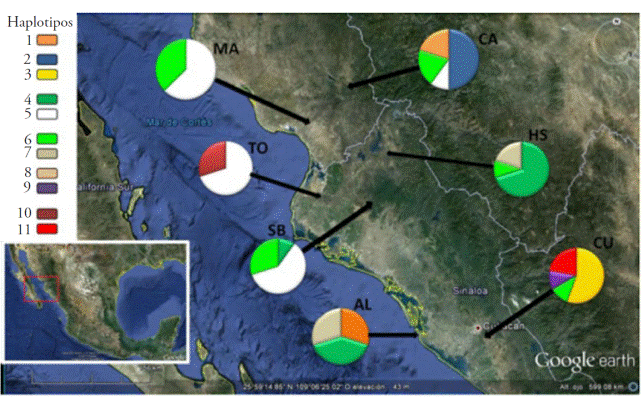
Figura 1 Distribución de haplotipos (sin considerar indels) de Pholisma culiacanum. Altata (AL), Culiacán (CU), Tosalibampo (TO), San Blas (SB), Heraclio SB (HS), Masiac (MA) y Cerro de Álamos (CA).
La diversidad nucleotídica total (π), sin considerar indels, fue 0.2737± 0.0242, donde la población con el valor mayor fue Masiaca (0.2990) y Tosalibampo (0.0007) con el menor. La diversidad haplotípica global, sin considerar indels (Hd), fue 0.855 ± 0.024, donde los valores mayores se compartieron entre Altata y Cerro de Álamos (0.7330) y el menor fue Tosalibampo (0.4670) (Cuadro 1).
Cuadro 1 Diversidad genética de Pholisma culiacanum.
| Localidad | h | Hd | hi | Hdi | π |
| Altata | 3 | 0.73300 | 10 | 1.00000 | 0.25957 |
| Culiacán | 4 | 0.72200 | 8 | 0.97222 | 0.12660 |
| Tosalibampo | 2 | 0.46700 | 9 | 0.97778 | 0.00069 |
| San Blas | 3 | 0.60000 | 9 | 0.97778 | 0.28244 |
| Heraclio SANB | 3 | 0.66700 | 6 | 0.84444 | 0.26463 |
| Masiaca | 2 | 0.53600 | 8 | 1.00000 | 0.29903 |
| Cerro de Alamos | 4 | 0.73300 | 10 | 1.00000 | 0.2960 |
| Total | 11 | 0.855 ± 0.024 | 57 | 0.99367 | 0.27366 ± 0.02425 |
Numero de haplotipos (h), diversidad haplotípica (Hd), número de haplotipos considerando indels (hi), diversidad haplotípica considerando indels (Hdi), diversidad nucleotídica (π), desviación estándar (±)
El valor de Fst (0.1349, p≤0.05) indica un nivel moderado de diferenciación genética entre las poblaciones estudiadas. El AMOVA mostró que la mayoría de la variación genética observada se debe principalmente a diferencias dentro de las poblaciones (86.51 %) y menos a las poblaciones (13.49 %). Este resultado coincide con los de otros estudios de plantas parásitas (Jerome et al., 2002; Amico et al., 2009) (Cuadro 2). Lo anterior podría esperarse pues los valores de Hd de las poblaciones son superiores a 0.5 (excepto Tosalibampo); niveles altos de varianza molecular dentro de poblaciones tienen como consecuencia diversidad haplotípica alta. El porcentaje alto de variación dentro de las poblaciones puede entenderse como consecuencia del flujo génico entre individuos de una misma población (García-Franco et al., 1998), esto contrasta con otros estudios de especies parásitas y dependientes, en las que gran parte de la variación genética se detectó entre poblaciones. Estas especies se caracterizan por su distribución fragmentada o en parches, por la dependencia de sus hospederos (de Vega et al., 2008), de la historia de vida tanto del parásito como del hospedero, de sufrir hechos geográficos y eventos estocásticos (cuellos de botella y evento fundador). Sin embargo, hay especies polinizadas por el viento que lo usan como medio de dispersión de semillas (como P. culiacanum), generalmente presentan índice de diversidad genética menor entre poblaciones (Allendorf et al., 2014).
Cuadro 2 Análisis de la varianza molecular.
| AMOVA Método de distancia: Tajima & Nei | ||||
| Variación | G.L | Suma de cuadrados | Varianza | Porciento de varianza |
| Entre poblaciones | 6 | 2247.257 | 23.44503 Va | 13.49 |
| Dentro de poblaciones | 60 | 9021.418 | 150.3569 Vb | 86.51 |
| Total | 66 | 11268.675 | 173.80201 | |
Índice de diferenciación genética Fst=0.13490 †Test de significancia 1023 permutaciones ᾽Va y FST: Valor de P= 0.0137±0.0034 §Weir, B.S. and Cockerham, C.C. 1984. Excoffier, L., Smouse, P., and Quattro, J. 1992. Weir, B. S., 1996.
Aunque hubo estructura poblacional con niveles bajos de diferenciación genética entre poblaciones, el coeficiente de correlación de Pearson y el modelo de regresión lineal de las distancias genéticas y geográficas no mostraron evidencia de aislamiento por distancia (p>0.05, r2=0.0026) a diferencia de otras plantas parásitas (Holzapfel et al., 2002; Jerome et al., 2002a; Mutikainen et al., 2002; deVega et al., 2008). Lo anterior permite inferir que existe flujo genético entre las poblaciones estudiadas y que éstas, en su conjunto, se comportan como una sola unidad evolutiva poblacional (García-Franco et al., 1998; Allendorf et al., 2014), la cual es tan grande que evita el efecto de la deriva génica. Sin embargo, en especies raras, como P. culiacanum, en las que la variación es igual o mayor que el de las especies comunes, se recomienda la evaluación de otros factores además de los ecológicos: las estrategias de ciclo de vida, los atributos reproductivos, la demografía, las interacciones con otros organismos (para este caso el hospedero) y otros (Eguiarte et al. 2007).
Distribución de haplotipos
El haplotipo 5 fue el de frecuencia mayor (19), se presentó en las poblaciones Tosalibampo, Masiaca, San Blas y Cerro de Álamos; el más distribuido fue el 6, que se presentó en Culiacán, San Blas, Heraclio Sb, Masiaca y Cerro de Álamos (Figura 2).
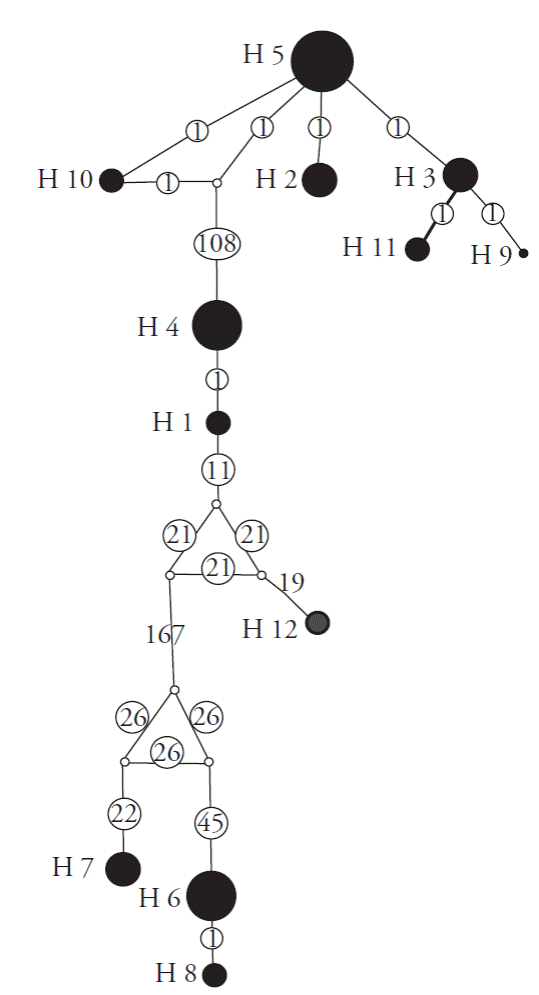
Figura 2 Red de haplotipos sin considerar indels, generada con el programa Networks (Bandelt et al., 1998). Las H y sus números corresponden a los haplotipos encontrados.
Las poblaciones con haplotipos únicos fueron Altata (haplotipo 1), Culiacán (haplotipos 3, 9, y 11), Tosalibampo (haplotipo 10) y Cerro de Álamos (haplotipos 2 y 8). Las otras poblaciones (Masiaca, San Blas y Heraclio Sb) estuvieron compuestas en su totalidad por haplotipos presentes en otras poblaciones (haplotipos 4, 5, 6 y 7). Los haplotipos no compartidos pueden ser consecuencia de mutaciones asociadas a eventos de colonización de nichos ecológicos nuevos (hospederos, ecosistemas o ambos). Esta capacidad de P.culiacanum para parasitar a diferentes géneros de plantas en diferentes ecosistemas fue documentada por Yatskievych (1985). También se puede deber a la expansión de los hospederos a nuevos territorios y la colonización posterior del parásito, como lo plantea deVega et al. (2008).
Red de haplotipos
En la Figura 2 cada haplotipo está representado por un círculo color negro cuyo tamaño es proporcional a la frecuencia de las secuencias que lo compone. En color blanco están representados los vectores medios que fueron generados por el programa para unir haplotipos distantes. Los vectores medios pueden ser pasos mutacionales, subespecies extintas o no muestreadas. El análisis detectó tres grupos grandes de haplotipos: 1) a 51 pasos mutacionales de Lennoa (H 12) se encuentran los haplotipos 1 y 4; 2) a 160 está un grupo formado por los haplotipos 2, 3, 5, 9, 10 y 11; 3) a 207 se encuentra otro grupo formado por los haplotipos 6, 7 y 8. Los métodos que utiliza la red de haplotipos están diseñados para modelar el pasado (evolutivo) mediante un proceso estocástico (coalescencia), el que está basado en el concepto de que los alelos en una población pueden rastrearse hacia atrás en el tiempo, hasta el punto en el que coalescen en un alelo ancestral. De esta manera, los haplotipos más lejanos a las secuencias raíz (6, 7, 8, 3, 11 y 9) son los más recientes, y los haplotipos (1 y 4) son los más antiguos (Eguiarte et al., 2007).
Filogenia de haplotipos
El árbol filogenético de los 11 haplotipos mostró cuatro grupos: 1) grupo externo (Lennoa); 2) los haplotipos 1 y 4, como el grupo basal de P. culiacanum y más cercano a Lennoa; 3) una rama que se bifurca en dos grandes grupos (80 % de soporte), por un lado los haplotipos cercanos a los basales 6, 8 y 7 (100 % de soporte); 4) una rama (100 % de soporte), en la que se encuentran los haplotipos 2, 3, 5, 9, 10, 11. La distribución general coincidió con la presentada en la red de haplotipos, incluida la cercanía o no de las secuencias raíz (Figura 3).
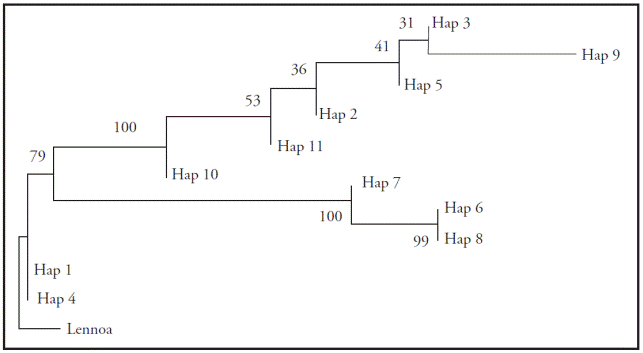
Figura 3 Filogenia de haplotipos de Pholisma culiacanum sin considerar indels utilizando el método Neighbor Joining, con el programa Mega (Tamura et al. 2007). Los valores de soporte de bootstrap son usando 1000 réplicas.
El aspecto morfológico de las poblaciones varía geográficamente. Dos grupos grandes se diferenciaron. El primero se formó con las poblaciones de Altata y Heraclio Sb y el segundo con las poblaciones de Culiacán, Tosalibampo, San Blas, Masiaca y Cerro de Álamos. El primero se caracterizó por tener tallos delgados únicos o ramificados que se originan en un haustorio único, con un aspecto parecido a los de Lennoa. El segundo grupo se caracterizó por tener tallos únicos, gruesos, con diámetro mayor a 1 cm. Estas observaciones visuales coincidieron con la evidencia molecular, producto de los análisis de filogenia y red de haplotipos que ubicaron a la mayoría de los haplotipos pertenecientes a las poblaciones del grupo uno, alejados de los del grupo dos.
En la red de haplotipos se observó gran cantidad de pasos mutacionales entre los haplotipos que en su mayoría están formados por individuos correspondientes a las poblaciones de Altata y Heraclio Sb (haplotipos 1, 4, 6, 7 y 8) y el resto de las poblaciones. La filogenia de haplotipos también ubicó a estos haplotipos de poblaciones, morfológicamente diferentes, cercanos entre sí y a Lennoa. Otros aspectos relevantes fueron la diferencia en el tipo de suelo donde se encuentran las poblaciones, dunas costeras (Altata) y suelo arenoso (Heraclio Sb); y suelos arcillosos de selva caducifolia, y los ecosistemas (selva espinosa, matorral xerófilo, selva subcaducifolia y selva caducifolia). Pholisma arenarium y Pholisma sonorae comparten estos tipos de suelos (Nabhan, 1979; Felger, 1980; Yatskievych, 1985; Yatskievych y Mason, 1986; AGFD Plant Abstract, 2004). Lo anterior permite sugerir que estas poblaciones de P. culiacanum pertenezcan a otra especie del genero Pholisma o a una sub especie de P. culiacanum. Por lo tanto, un análisis molecular comparativo entre las tres especies del género es necesario para determinar la razón de estas diferencias.
Dados los niveles altos de diversidad genética haplotípica es recomendable establecer zonas de conservación de recursos genéticos en las áreas de distribución de las poblaciones de Culiacán, Cerro de Álamos y Altata. Éstas abarcarían casi la totalidad de la diversidad genética de las poblaciones estudiadas y la mayoría de los haplotipos únicos quedarían dentro de las áreas conservadas. Esto es importante porque estas áreas de distribución son las más afectadas por actividades antropogénicas, como el desarrollo urbano y turístico, la agricultura, la tala de bosque primario y el cambio de uso de suelo.
Con fines de explotación in situ, se recomienda la zona de distribución Tosalibampo porque los índices de diversidad genética y haplotípica fueron los menores, contó únicamente con dos haplotipos y fue la población más abundante.

