











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
El cáncer colorrectal es la tercera causa de cáncer y la cuarta causa más común de muertes relacionadas por cáncer en el mundo. La mayoría se presenta de forma esporádica, de 70 a 80% aproximadamente y de 10 a 20% presentan relación familiar.1
La poliposis adenomatosa familiar (PAF) es un trastorno hereditario autosómico dominante que aparece en aproximadamente 1:10,000 nacimientos vivos y afecta ambos géneros por igual y a todas las razas. Puede ser asintomático o bien presentarse con sangrado, diarrea, dolor abdominal o descarga mucosa. También se presenta con anemia, oclusión intestinal o pérdida de peso cuando hay presencia de pólipos de gran tamaño o aumentan su número precediendo al desarrollo de cáncer. La principal característica es la presencia de cientos a miles de adenomas colorrectales con zonas de mucosa normal entre cada lesión. La poliposis leve es aquella donde se identifican entre 100 y 1,000 adenomas. Cuando se encuentran menos de 100 adenomas recibe el diagnóstico de poliposis atenuada. Cerca de 100% de los pacientes desarrollarán cáncer si no reciben tratamiento.2
La PAF es una enfermedad multisistémica que puede presentar numerosas manifestaciones extracolónicas. Éstas incluyen adenomas gastroduodenales y cáncer, tumores desmoides, osteomas, quistes epidermoides, cáncer papilar de tiroides, pólipos del intestino delgado y cáncer, hiperplasia congénita del epitelio pigmentario de la retina, y anomalías dentales.
El síndrome de Gardner es una poliposis que se acompaña de tumores desmoides, osteomas, quistes epidermoides o dientes supernumerarios. El síndrome de Turcot es una PAF asociada a tumores malignos del sistema nervioso central. Ambos síndromes son causados por mutaciones en el gen APC (adenomatous polyposis coli).
Bases genéticas
La PAF es ocasionada por una mutación hereditaria en el gen APC ubicado en el cromosoma 5q21.2,3 Los pacientes afectados nacen con una sola copia funcional de este gen “guardián”. La pérdida del segundo alelo conduce al rápido desarrollo de cientos a miles de adenomas colorrectales. Se han descrito más de 850 mutaciones distintas, en las cuales hay un desarrollo anómalo de la proteína APC. Cerca de 25% de los pacientes con PAF tiene una mutación de novo, sin ningún antecedente familiar.4
Diagnóstico
La poliposis adenomatosa familiar se puede diagnosticar genética o clínicamente. Las pruebas genéticas revelan una mutación del gen APC en aproximadamente 80% de los casos. Las indicaciones para referencia a consejo genético incluyen historia familiar de PAF, historia personal de 10 o más adenomas, historia personal de adenomas, y manifestaciones extracolónicas de la enfermedad. En los pacientes en riesgo con familiares con mutación conocida se realizan las pruebas dirigidas para dicha mutación. En aproximadamente 20% de los pacientes no se puede identificar la mutación, pero el fenotipo clínico está presente.5
Riesgo de desarrollo de cáncer
La PAF presenta un riesgo de desarrollar cáncer colorrectal cercano a 100%. Representa sólo de 0.5 a 1% de las causas de cáncer colorrectal y en promedio se presenta a los 39 años de edad. El objetivo del tratamiento en estos casos es aumentar la supervivencia disminuyendo el riesgo de muerte por cáncer colorrectal realizando colectomía o proctocolectomía antes de que el cáncer se presente. El riesgo de cáncer en la PAF atenuada es de 70% aproximadamente, y se desarrolla a una edad mayor comparado con la enfermedad clásica.5,6
Manifestaciones extracolónicas de la poliposis adenomatosa familiar
Aproximadamente 90% de los pacientes con PAF desarrollan adenomas duodenales, pero sólo 5-10% desarrollan cáncer periampular. Hasta en 50% de los pacientes se encuentran pólipos en el fondo gástrico, los cuales tienen mínimo riesgo de malignidad.5
La enfermedad desmoide afecta a aproximadamente 5% de los pacientes con poliposis. Cerca de la mitad de los tumores se originan en el mesenterio y 40% se desarrollan en la pared abdominal. El resto se presenta en la espalda, cuello o extremidades. Los tumores desmoides se manifiestan como lesiones planas, fibrosas o masas discretas. Se asocian más frecuentemente al género femenino e historia familiar de tumores desmoides. La mayoría de estas lesiones se desarrollan cinco años después de una cirugía abdominal posiblemente como parte de la respuesta inflamatoria.5
El riesgo de presentar cáncer tiroideo es de sólo 2%; sin embargo, corresponde al doble de la población general. La incidencia es 17 veces mayor en mujeres que en hombres y se desarrolla en promedio a los 27 años de edad. La histología principal es carcinoma papilar.5
Entre las lesiones malignas asociadas menos frecuentes están el adenocarcinoma pancreático, hepatoblastoma y meduloblastoma. Existen también lesiones benignas asociadas que si bien no requieren de tratamiento, son útiles para el diagnóstico. La hipertrofia congénita del epitelio pigmentario de la retina corresponde a lesiones ovales gris oscuro o marrones observadas en 60-85% de los pacientes. Los osteomas son anormalidades dentales que se observan en 20% de los casos. Se pueden apreciar algunas lesiones cutáneas como quistes dermoides, lipomas o fibromas. Su presencia en la cara, cuello, cráneo o extremidades suelen sugerir el diagnóstico de PAF.5,7
Estudios de tamizaje para poliposis adenomatosa familiar
Se deben realizar estudios de tamizaje tanto para la enfermedad colónica como para las manifestaciones extracolónicas con el objetivo de limitar el riesgo de desarrollar cáncer realizando una intervención oportuna y referencia quirúrgica. El tamizaje debe hacerse en cada individuo mediante diagnóstico genético o en familiares de primer grado con diagnóstico de PAF. Aun con sólo el diagnóstico clínico se debe hacer un estudio completo de los familiares. Los estudios deben comenzar a los 12 años de edad mediante rectosigmoidoscopia flexible. Si se observan pólipos se debe solicitar colonoscopia completa. Si no existen pólipos en el examen inicial, la sigmoidoscopia deberá repetirse cada uno a dos años o antes si se presentan síntomas.5
Deberá realizarse la primera panendoscopia entre los 20 y 25 años de edad y los intervalos de vigilancia se realizan de acuerdo a los hallazgos. En el caso de enfermedad tiroidea, cada año deberá realizarse exploración física y ultrasonido en busca de lesiones sospechosas. Para tumores desmoides y otras lesiones extracolónicas no existe un sistema de tamizaje específico; sin embargo, deberán realizarse estudios especiales en caso de que exista alta penetración de alguna lesión particular en otros miembros de la familia.7,8
Tratamiento
Los objetivos del tratamiento en la poliposis adenomatosa familiar son eliminar o limitar el riesgo de cáncer e incrementar tiempo de vida y mejorar la calidad de vida del paciente. Ya que la posibilidad de desarrollar cáncer es alta, el tratamiento quirúrgico oportuno es la base del tratamiento. La decisión sobre el tiempo de la cirugía depende de la presencia de síntomas, la edad del paciente al momento del diagnóstico y otras circunstancias especiales. Cuando existe sintomatología se debe ofrecer al paciente cirugía para tratar los propios síntomas y de forma profiláctica por el potencial riesgo de presentar cáncer. En los niños y adolescentes asintomáticos la cirugía se retrasa de forma razonable hasta el final de la adolescencia o alrededor de los 20 años, cuando se ha alcanzado cierta madurez física y emocional. Debido a que el riesgo de cáncer aumenta con la edad, a partir de la tercera década de la vida se ofrece el tratamiento quirúrgico a los pacientes en el momento del diagnóstico.7
Para los pacientes en quienes no hay evidencia de cáncer rectal se puede ofrecer colectomía con anastomosis ileorrectal o proctocolectomía. Esta última elimina toda la mucosa de riesgo y la posibilidad de desarrollar cáncer en el futuro.9 La íleo-ano anastomosis en bolsa o reservorio ileal (pouch) presenta mayor movimiento intestinal, mayor riesgo de incontinencia y se ve afectada la calidad de vida en comparación con la anastomosis ileorrectal; sin embargo, el riesgo de desarrollar cáncer en la mucosa rectal remanente es de 4-10%. El riesgo de desarrollar cáncer aumenta hasta 12% después de 20 años de la cirugía. 42% de los pacientes postoperados de anastomosis ileorrectal requerirán en un futuro de proctectomía por presencia de cáncer o poliposis descontrolada.9
Cuando existe cáncer de colon y metástasis a distancia la decisión sobre el tipo de cirugía se basa en la posibilidad, aunque sea baja, de que exista un cáncer metacrónico en el recto si éste se respeta. Pacientes con tumores localmente avanzados (o que existe riesgo de metástasis) con presencia mínima de pólipos se beneficiarán de una colectomía abierta o por vía laparoscópica individualizando cada caso, con anastomosis ileorrectal o bien proctocolectomía con ileostomía terminal. En pacientes con cáncer en estadio IV con baja expectativa de vida se recomienda proctectomía si no existe cáncer de colon o la presencia de pólipos es mínima. Debe tomarse en cuenta que la proctectomía está asociada a un mayor riesgo de disfunción urinaria y sexual, disminución de la fertilidad en las mujeres y menor calidad de vida en general.10
Un porcentaje pequeño de pacientes desarrolla cáncer en la zona de transición anal o en la zona de anastomosis del reservorio ileal. Se ha propuesto la mucosectomía como alternativa, pero si ésta no se realiza de forma adecuada puede fallar en su propósito. Hasta 21% de los pacientes a quienes se realiza mucosectomía presentan siembras de cáncer cerca de las bolsas ileales.8
El adenoma duodenal puede progresar a cáncer de forma poco frecuente (hasta 11%) y las lesiones pueden ser tratadas mediante endoscopia alta.7
Los tumores desmoides se presentan desde asintomáticos hasta con dolor severo, oclusión intestinal o con fístula. El tratamiento depende de los síntomas, localización, tamaño y extensión de la enfermedad.
Los nódulos tiroideos mayores de 1 cm deben analizarse mediante punción con aguja fina. El cáncer de tiroides se trata mediante tiroidectomía total y ablación con yodo.
Evaluación de familiares en riesgo
Todos los familiares en primer grado tienen 50% de posibilidades de desarrollar PAF, por lo que deben ser evaluados inicialmente por un especialista en genética. Los familiares potencialmente afectados deben estudiarse al momento del diagnóstico y en el caso de los niños al cumplir los 12 años de edad. Cuando se conoce una mutación del APC en la familia lo más recomendable es una prueba de ADN. Los familiares en riesgo que no cuentan con diagnóstico genético deben iniciar la vigilancia a los 12 años de edad con sigmoidoscopia flexible o bien, colonoscopia si el tamizaje se inicia en la edad adulta. Los familiares que no presentan pólipos a los 40 años de edad se manejarán según las guías para la población general.7
Caso clínico
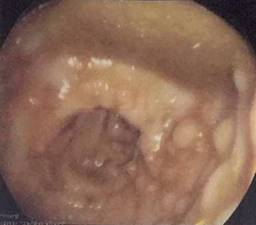
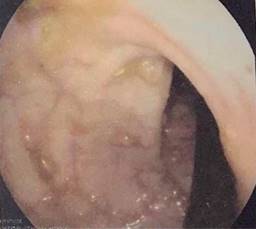
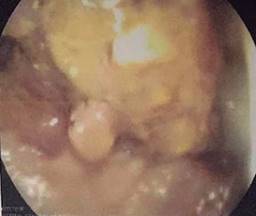
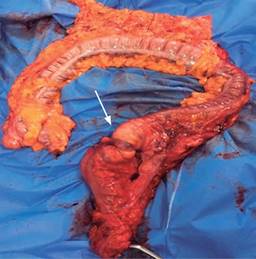
Paciente femenino de 48 años de edad que inició con hematoquecia hace seis meses aproximadamente, pérdida de más de 10 kilogramos y dolor abdominal inespecífico. Presenta también antecedentes de tres familiares en primer grado con diagnóstico de cáncer de colon y un familiar en segundo grado con diagnóstico de poliposis. Se realiza colonoscopia encontrando más de 100 pólipos sésiles y a 25 cm del margen anal una tumoración estenosante (Figuras 1 y 2). Se somete a la paciente a proctocolectomía abierta con ileostomía terminal identificando tumor a nivel de colon sigmoides de 3 × 2 cm (Figuras 3 y 4). El estudio histopatológico reporta adenocarcinoma moderadamente diferenciado invasor de colon. Se clasificó como un T4a N1b M0, estadio IIIB, por lo que recibió tratamiento adyuvante con quimioterapia.
Discusión
La poliposis adenomatosa familiar es un padecimiento poco frecuente que requiere de un diagnóstico oportuno y preciso para prevenir la formación de cáncer colorrectal tanto en el paciente como en los familiares de primer grado. La cirugía sigue siendo el único tratamiento preventivo en la actualidad; la proctocolectomía, con o sin restitución del tránsito intestinal, es el mejor procedimiento, pues elimina la mucosa de riesgo de formación de cáncer.

