










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkCirujano general
versión impresa ISSN 1405-0099
Cir. gen vol.34 no.4 Ciudad de México oct./dic. 2012
CASO CLÍNICO
Tumores neuroendocrinos de páncreas debutando como ampuloma: reporte de un caso y revisión del abordaje diagnóstico actual
Neuroendocrine tumors of the pancreas appearing as ampulomas: report of one case and review of the current diagnostic approach
Josué Alejandro Andrade-Bucio, Alan Fernando Andrade-Bucio, Edgar Uriel Hernández-Velázquez, Ariadne Beltrán-Estrada, Fernando Rodríguez-Ortega, Arturo Morales-Padilla, César Jaramillo-Martínez
Departamentos de Cirugía General y Anatomía Patológica, Centro Médico ISSEMYM.
Correspondencia:
Dr. Andrade Bucio Josué Alejandro
Centro Médico del Instituto de Seguridad Social del Estado de México y Municipios.
Departamento de Cirugía General.
Av. Baja velocidad Núm. 284, San Jerónimo Chicahualco, Metepec, Estado de México, 52170.
Tel: (01 722) 2756300, ext. 2156, 2160; fax: 2756300.
E-mail: fro2411@hotmail.com
Recibido para publicación: 27 septiembre 2011
Aceptado para publicación: 12 febrero 2012
Resumen
Objetivo: Demostrar un ejercicio clínico-quirúrgico del abordaje diagnóstico actual para el tratamiento de tumor neuroendocrino de páncreas con resultados satisfactorios en un paciente seleccionado.
Sede: Centro Médico ISSEMYM (Tercer nivel de atención).
Diseño: Reporte de caso.
Descripción del caso: Mujer de 42 años con diagnóstico de pancreatitis aguda, las bilirrubinas mostraban patrón obstructivo. Se realizó colangiopancreatografía endoscópica retrógrada mostrando imagen sugestiva de ampuloma ulcerado; el reporte histopatológico refirió: adenocarcinoma pobremente diferenciado. En el curso de su evolución presentó colangitis, que requirió tratamiento con nueva realización de colangiopancreatografía endoscópica retrógrada con esfinterotomía, aseo coledocal con balón y colocación de endoprótesis plástica 10 French, con buena respuesta clínica. Se programó pancreatoduodenectomía, encontrando tumoración en proceso uncinado de páncreas y tumor en segunda porción de duodeno, cuyo reporte transoperatorio fue tumor neuroendocrino.
Conclusión: Los tumores neuroendocrinos de páncreas son neoplasias poco comunes, cuya incidencia reportada en la literatura especializada es <1 por cada 100,000 personas por año. Provienen de células pluripotenciales dentro del páncreas exocrino y comprenden <2% de todos los tumores pancreáticos, son capaces de producir hormonas (insulina, gastrina, VIP, etcétera) y su tratamiento es primordialmente la resección quirúrgica. A pesar de tratarse de lesiones infrecuentes, los tumores neuroendocrinos de páncreas deben estar en la mente del cirujano general ante cuadros de colangitis de repetición o como probabilidad diagnóstica de patología biliar de corte maligno, sugerida por una evolución tórpida y apoyada con estudios de gabinete sensibles a su diagnóstico.
Palabras clave: Tumor neuroendocrino, páncreas, ampuloma, pancreatoduodenectomía.
Abstract
Objective: To demonstrate a clinical-surgical exercise of the current diagnostic approach for the treatment of a neuroendocrine tumor of the pancreas, with satisfactory results in a selected patient.
Setting: Centro Médico ISSEMYM (Third level health care).
Design: Case report.
Description of the case: A 42-year-old woman with diagnosis of acute pancreatitis, bilirubins indicated an obstructive pattern, retrograde endoscopic cholangiopancreatography was performed that revealed an image suggestive of ulcerated ampuloma; the histopathological report referred: poorly differentiated adenocarcinoma. During evolution, the patient presented cholangitis, which required treatment and a new retrograde endoscopic cholangiopancreatography with esphyncterotomy, choledochus cleaning with balloon, and placement of a plastic French 10 endoprosthesis, with good clinical response. Pancreatoduodenectomy was programmed, finding a pancreatic uncinate process tumor and a tumor in the second portion of the duodenum, the transoperative report corresponded to a neuroendocrine tumor.
Conclusion: Neuroendocrine tumors of the pancreas are rare neoplasms, with a literature reported incidence of less than 1 per 100,000 people per year. They originate from pluripotent cells within the exocrine pancreas and constitute <2% of all pancreatic tumors. They are able to produce hormones (insulin, gastrin, VIP, etc.) and their treatment is mainly surgical. Despite being infrequent tumors, neuroendocrine tumors of the pancreas must always be present in the mind of the general surgeon when faced with repeated cholangitis symptoms or as a diagnostic probability of malignant type biliary pathology as suggested by a torpid evolution and supported by imaging studies sensitive to the diagnosis.
Key words: Neuroendocrine tumor, pancreas, ampuloma, pancreatoduodenectomy.
Introducción
Los tumores neuroendocrinos de páncreas (TNEP) son neoplasias poco comunes, cuya incidencia reportada en la literatura especializada es <1 por cada 100,000 personas por año. Provienen de células pluripotenciales dentro del páncreas exocrino y comprenden <2% de todos los tumores pancreáticos.1,2
Los TNEP pueden derivarse de islotes de Langherhans, de células neuroendocrinas del epitelio respiratorio y células parafoliculares del tiroides (su diagnóstico es más común post mortem en 0.8 a 10% de las autopsias), también comparten propiedades citoquímicas similares con tumores carcinoides, con ciertas neoplasias de la glándula pituitaria, paratiroides y médula adrenal. El término neuroendocrino confiere una mejor definición que el término carcinoide (aunque este último continúa siendo usado para referirse a tumores que secretan serotonina) y se refiere a que este grupo de neoplasias comparten antígenos con las células nerviosas, produciendo sustancias características de diferenciación neuronal, como enolasa neuroespecífica, cromogranina A, B, C y sinaptofisina, lo que ofrece una sensibilidad elevada en su diagnóstico al valorar estas sustancias como marcadores tumorales.1,3 Su etiología es pobremente entendida, en su mayoría son espontáneos, con una leve carga familiar para su predisposición.
Son más comunes entre la quinta y sexta décadas de la vida, de localización más frecuente en la cabeza del páncreas. Al momento del diagnóstico, un tercio de los pacientes cursa con ictericia. Setenta al 90% son malignos y la mayoría cursan con metástasis, generalmente en el hígado y rara vez en los huesos. Son difíciles de diferenciar de los tumores exocrinos de páncreas, teniendo un rasgo especial, que a diferencia de los tumores exocrinos de páncreas los TNEP no invaden estructuras vasculares ni la vía biliar.1,4-6
Se constituyen en un grupo heterogéneo de neoplasias que al originarse de células neuroendocrinas (distribuidas difusamente a lo largo del intestino), con propiedades secretoras, confieren a aproximadamente el 60% de estos tumores, la particularidad de secretar uno o dos péptidos biológicamente activos; los más frecuentes son: insulinoma, gastrinoma, glucoganoma, VIPoma y somatostatinoma, dando lugar a síndromes hipersecretores, incluido el síndrome carcinoide.1,3,6,7
Sin embargo, del 15 al 30% de los tumores neuroendocrinos del páncreas son no funcionales, éstos generalmente son diagnosticados por los síntomas causados por el efecto de masa que ejerce el tumor sobre estructuras adyacentes.1,6,8
Los TNEP no funcionales pueden tener un curso indolente o presentarse con síntomas obstructivos (dolor, náusea y vómito), mientras que las manifestaciones clínicas de los TNEP funcionales son más floridas; el cuadro I resume los síntomas típicamente encontrados en aquellos pacientes con síndromes secretores.3,9
El abordaje diagnóstico actual se encuentra cimentado en evidencias clínicas de imagen, de laboratorio y de histopatología, lo que orientará al cirujano a tomar una decisión terapéutica, para la enfermedad maligna, la enfermedad resecable y para la que no lo es.
La resección quirúrgica completa de los TNEP continúa siendo la única esperanza de curación. La presencia de invasión vascular y perineural, extensión linfática y metástasis hepáticas impactan su pronóstico.1
Los TNEP tienen, en general, un pronóstico más alentador que las neoplasias ductales más comunes del páncreas,7 cursan en su mayoría debajo del horizonte clínico aun tratándose de enfermedad metastásica. La causa más común de muerte en estos pacientes es la falla hepática.
Presentación del caso
Mujer de 42 años con rama paterna positiva para cáncer de próstata, inicia su padecimiento 14 días previos a su ingreso al Centro Médico con presencia de dolor abdominal en hipocondrio derecho tipo cólico leve, intermitente, que gradualmente aumentó de intensidad 7/10, sin horario ni predominio; como síntomas acompañantes, náusea y vómito de características gastrobiliares, la ingesta de colecistoquinéticos predisponían a presentar la sintomatología. Negaba pérdida de peso. A la exploración física destacaban hiperbaralgesia en hipocondrio derecho e interrupción de la inspiración pronunciada a la palpación profunda de cuadrante superior derecho (signo de Murphy positivo). Se solicitaron pruebas de laboratorios a su ingreso: leucocitos (LEUC) 4.85; hemoglobina (Hb) 12.9; hematócrito (Hcto) 36.9; plaquetas 238; glucosa (Gluc) 61; urea 22; creatinina (Creat) 0.60; calcio (Ca) 7.34; lactato deshidrogenasa (LDH) 377; alanino aminotrasferasa (ALT) 136; aspartato aminotrasferasa (AST) 269; bilirrubinas totales (BT) 1.23; bilirrubina directa (BD) 1.13; bilirrubina indirecta (BI) 0.10; fosfatasa alcalina (FA) 355; albúmina 3.6; amilasa 453; lipasa 466; colesterol 164; triglicéridos 74. Compatible clínicamente, y por resultados de laboratorio, con un cuadro de pancreatitis aguda con patrón biliar.
Como parte del protocolo de estudio para esta patología se solicitó ultrasonografía (USG) de hígado y vías biliares, reportándose: colédoco de 12.4 mm, presencia de líquido perivesicular, pared de vesícula de 3.6 mm, vesícula biliar de 113.9 x 48 x 51.1 mm, conducto de Wirsung de 4.7 mm. El diagnóstico radiológico presuntivo fue de dilatación de vía biliar intrahepática, coledocoectasia, ectasia de conducto de Wirsung y colecistitis alitiásica.
Al no documentarse litos como causa de la obstrucción de vías biliares, se complementó protocolo de estudio con una colangiorresonancia magnética (CRM) en búsqueda de la etiología del patrón obstructivo que la paciente presentaba; reportando dilatación de la vía biliar intrahepática y extrahepática (Figura 1) y del conducto de Wirsung, con imagen de defecto de llenado, lo que se traduce como una imagen sugestiva de obstrucción.

Por documentarse dos imágenes sugestivas de obstrucción, obtenidas por USG y CRM, se decidió realizar colangiopancreatografía endoscópica retrógrada (CPRE), la cual fue fallida, debido a imposibilidad para canular el ámpula duodenal porque presentaba una lesión ulcerativa de 3 x 3 cm, con estigmas de sangrado reciente y bordes peripapilares infiltrados. Se tomaron biopsias y, ante el hallazgo de la tumoración ampular, se solicitaron y obtuvieron títulos altos para alfafetoproteína de 247.9 mg/dl (rango normal 0-5.1 mg/dl) y CA 19.9 de 43.7 mg/dl (rango normal de 0-37 mg/dl).
El resultado histopatológico de la tumoración fue: adenocarcinoma pobremente diferenciado ulcerado (Figura 2)

Una vez obtenido el reporte de adenocarcinoma y con la intención de descartar enfermedad metastásica, al momento del diagnóstico se realizó tomografía computada (TC), la cual reportó signo de doble conducto, sin evidencia de enfermedad metastásica, sólo lesión en luz duodenal.
Siete días posteriores a su ingreso, la paciente presentó dolor en hipocondrio derecho, intensidad 10/10, fiebre de 39 grados e ictericia (tríada de Charcot), así como hiperbaralgesia en epigastrio e hipocondrio derecho, por lo que se solicitaron nuevos estudios de laboratorio: LEUC 9.17; ALT 8.9; AST 150.6; BT 8.31; BD 7.47; BI 0.84; FA 568; gamma-glutamil transpeptidasa (GGT) 877; LDH 313; CA 8.11; amilasa 164; lipasa 19.
Por cuadro clínico y laboratorio se documentó episodio de colangitis, cuyo tratamiento inicial consistió en realización de nueva CPRE, reportando tumor ampular menor de 3 cm, colédoco de 18 mm, realización de esfinterotomía, limpieza coledocal con balón y colocación de endoprótesis plástica 10 French. Posterior al procedimiento y cobertura con triple esquema antibiótico, la evolución de la paciente mostró buena respuesta clínica, hasta que los síntomas remitieron.
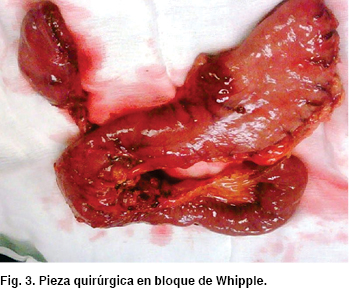
En presencia de tumoración de ámpula duodenal (ampuloma) sin hallazgos de enfermedad metastásica, descartados por TC, pero mayor a 1 cm de tamaño, lo dejaba fuera del rango aceptado para la terapéutica de resección endoscópica, por lo que se optó por el tratamiento quirúrgico, sometiendo a la paciente a pancreatoduodenectomía o procedimiento de Whipple (Figura 3)
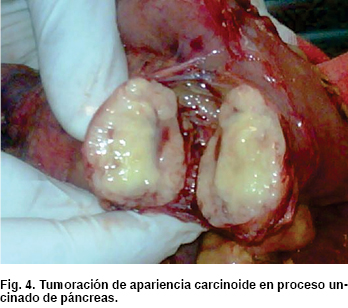
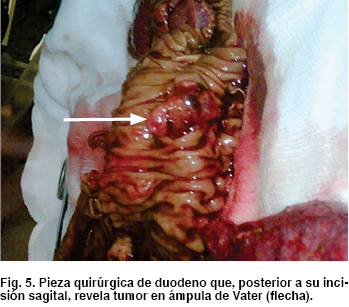
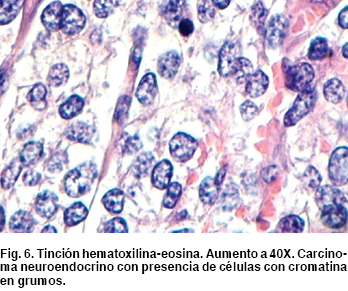
Como hallazgos transquirúrgicos se encontró tumoración a nivel de proceso uncinado de páncreas de 3 x 3 cm (Figura 4) Se resecó ganglio retrocoledociano y se observó un segundo tumor en tercera porción de duodeno de 2 x 2 cm (Figura 5), cuyo reporte histopatológico transoperatorio (Figura 6) fue tumor neuroendocrino pobremente diferenciado de ámpula de Vater y cabeza de páncreas, tres ganglios linfáticos con metástasis de tumor neuroendocrino y límites quirúrgicos libres.
La evolución de la paciente en el postquirúrgico fue lenta pero consistente hacia su mejoría clínica total. La paciente fue dada de alta 13 días posteriores a la realización del procedimiento de Whipple. Con leve palidez de tegumentos, heridas quirúrgicas adecuadamente afrontadas, sin evidencia de hematoma, seroma o infección. Laboratorios a su egreso: LEUC 10.87; Hb 10.4; Hcto 31.6; Gluc 112; urea 24; Creat 0.43; ALT 16.5; AST 35.7; BT 0.53; BD 0.40; BI 0.13; FA 116. Tomografía de control con cambios postquirúrgicos sin evidencia de colecciones.
El estudio inmunohistoquímico al que se sometió la pieza quirúrgica, como complementación, reportó carcinoma endocrino de células claras del páncreas y carcinoide clásico de ámpula de Vater.
Al momento actual la paciente se encuentra en seguimiento por la consulta externa de cirugía general y oncológica clínica, sin datos de enfermedad activa, sin presencia de enfermedad metastásica, corroborado por TC de control y Octreoscan negativo, incorporándose a sus actividades cotidianas.
Discusión
El caso médico-quirúrgico que aquí se expuso ejemplifica un escenario clínico de un TNEP no funcional, por lo que enfocaremos nuestra atención al diagnóstico diferencial y tratamiento quirúrgico entre los TNEP no funcionales y los TNEP que, debido a la sobreproducción hormonal, causan síndromes hipersecretores específicos.
Los TNEP no funcionales deben ser sospechados por los síntomas obstructivos (dolor, ictericia, vómito) que resultan del efecto de masa que el tumor ejerce directamente sobre las estructuras adyacentes a él (ámpula de Vater, cístico, colédoco, hígado, vena porta, vasos pancreato-esplenomesentéricos ), ya que no producen sintomatología por la sobreproducción hormonal o porque ésta no es lo suficientemente importante para tener relevancia clínica. Hasta en el 16% de los casos el tumor se descubre de forma incidental.1,4,8,9
Ante los síntomas de obstrucción, por el efecto de masa que el tumor ejerce sobre estructuras adyacentes, y ante una alta suspicacia clínica de que pueda tratarse de TNEP, se deben analizar las siguientes situaciones: 1) ¿Se tratará de TNEP no funcionante o funcionante? En caso de que las manifestaciones clínicas permitan al clínico sospechar de este último, los síntomas estarían más cercanos a constituir un síndrome hipersecretor (Cuadro I). Pero al no existir sintomatología compatible con tales síndromes, pueden descartarse clínicamente. Por lo tanto, las determinaciones de péptidos y hormonas en orina, producto de la degradación de serotonina (presentes en TNEP funcionales), estarían erróneamente indicadas por el cirujano. Ante esta evidencia clínica nosotros reservaríamos este recurso de laboratorio y optaríamos por la determinación en sangre de marcadores tumorales o pruebas de funcionamiento hepático. 2) Se debe contar con al menos dos criterios de cuatro posibles para sustentar el diagnóstico de TNEP (el cuadro II despliega cada uno de los cuatro criterios). 3) Es necesario, para el diagnóstico definitivo de TNEP, el estudio histopatológico-inmunohistoquímico, ya que es el eslabón final de los esfuerzos enmendados en el abordaje diagnóstico.
En el caso presentado ejemplificamos un abordaje diagnóstico, el cual se fundamentó en los cuatro criterios arriba mencionados. Estos son: criterios clínicos, de laboratorio, de imagen y los histopatológicos. La presencia de al menos dos de cuatro de estos criterios será altamente sugestiva de diagnóstico de tumor neuroendocrino de páncreas; aunque la especificidad y sensibilidad para TNEP cambia de un estudio a otro, el porcentaje de certeza es mayor si se solicitan como estudios combinados que como estudios aislados.
1) Criterio clínico: Síntomas obstructivos (dolor, ictericia, vómito) que a lo largo de la estancia hospitalaria de la paciente constituyeron entidades patológicas bien definidas, como lo son pancreatitis aguda/colangitis (el cuadro I resume los síntomas típicamente encontrados en aquellos pacientes con TNEP que producen síndromes secretores).
2) Criterio de imagen: Cuando se sospecha un TNEP, un estudio preoperatorio de imagen para la localización del mismo está indicado, así como para descartar metástasis. La angiografía y la ultrasonografía de alta resolución pueden proveer información útil, aunque en la mayoría de los casos los hallazgos son inespecíficos. La TC simple con contraste oral e intravenoso del abdomen y de la pelvis, así como la resonancia magnética (RM) del hígado y del páncreas, permiten detectar el tumor primario y la enfermedad metastásica del hígado y de los ganglios linfáticos. Los TNEP expresan una alta densidad de subtipos de receptores somatostatínicos con capacidad de unión a los análogos de la somatostatina, por lo que la gammagrafía de los receptores somatostatínicos con indio-111 es, hoy por hoy, la prueba más sensible para localizar la mayoría de los TNEP.10,11 Las lesiones accesibles por endoscopia pueden ser mejor definidas por ultrasonido endoscópico (UE), delimitando la invasión local y hacia cadenas linfáticas regionales. La tomografía por emisión de positrones (PET) es otra técnica actual que cuando se combina con el 5-hidroxitriptofano marcado con 11C identifica y localiza anatómicamente más del 95% de los TNEP.12-15
En el caso presentado durante el diagnóstico y tratamiento convencional de la pancreatitis/colangitis que presentó la paciente, el estudio de USG, CRM y el endoscópico (CPRE) nos aportaron dicha evidencia al visualizar una vía biliar intrahepática y extrahepática dilatada, en continuidad con una ámpula de Vater con tumoración ulcerada de 3 x 3, de la cual se tomaron biopsias.
3) Criterio de laboratorio: Se solicitaron (ante el hallazgo de probable ampuloma reportado en la CPRE) marcadores tumorales, alfafetoproteína de 247.9 mg/dl (rango normal 0-5.1 mg/dl) y CA 19.9 de 43.7 mg/dl (rango normal de 0-37 mg/dl), con títulos altos pero inespecíficos, que se explican por la obstrucción de la vía biliar que en ese momento presentaba la paciente y de la cual se obtuvo evidencia bioquímica constituida en la elevación de las enzimas hepatobiliares y por el patrón obstructivo que denotaban las bilirrubinas elevadas. Títulos altos del antígeno carbohidratado (CA) 19-9 y del antígeno carcinoembrionario (ACE) se observan en el 50-79%16 y del 40-70%,17 respectivamente, en los pacientes con indicios clínicos de malignidad en la vía biliar.18 En el cuadro II se enumeran los marcadores bioquímicos específicos para TNEP funcionales.
4) Criterio histopatológico: Se demostró una vez obtenido el resultado del estudio inmunohistoquímico de la pieza quirúrgica resecada por cirugía, que reportó: carcinoma endocrino de células claras del páncreas y carcinoide clásico de ámpula de Vater. Algunos rasgos anatomopatológicos de los TNEP, que indican malignidad, son el tamaño tumoral, invasión de la submucosa del tejido adyacente, signos de invasión vascular o de los espacios perineurales, atipia estructural con prevalencia de áreas sólidas amplias, atipia celular, más de dos mitosis por 10 campos de gran aumento, presencia de necrosis, acumulación nuclear de p53, aumento del número de núcleos con positividad para Ki-67 y pérdida de la cromogranina A (desdiferenciación celular).19
De lo anterior hay que destacar que el interrogatorio y la exploración clínica continuarán a través de la revolución vertiginosa de la medicina moderna, siendo la directriz insustituible y más importante para sospechar el origen de una enfermedad y así solicitar intencionadamente estudios paraclínicos que sustenten la etiología de la patología, y no en el sentido inverso. El estudio histopatológico es el eslabón final de los esfuerzos enmendados en el abordaje diagnóstico, ya que es muy improbable distinguir -por lo que el cirujano ve y toca en el acto quirúrgico- estos tumores de otros en el preoperatorio y transoperatorio.
Como se identificó en este caso clínico, el tratamiento quirúrgico constituye el patrón de oro para los TNEP no funcionales, pues a diferencia de la mayoría de tumores endocrinos del páncreas, en éstos debe intentarse la resección pancreática formal. Los tumores neuroendocrinos de páncreas y periampulares forman un grupo especial que requiere particular atención; en primer lugar, lo óptimo es que el diagnóstico pueda establecerse previo a la cirugía, la evidencia de laboratorio puede orientar sobre el sitio del tumor (por ejemplo, el triángulo del carcinoma) y la probabilidad de malignidad (por ejemplo, baja en el insulinoma).3 Si la lesión es localizable por criterio de imagen previo a la cirugía y está bien definida y cercana a la superficie del páncreas, la enucleación puede ser suficiente, comprobando, mediante criterio histopatológico, escisión completa y rasgos benignos de la lesión. Si esto no es posible estará justificado realizar una pancreatoduodenectomía de Kausch-Whipple o pancreatectomía distal de Duval, en casos seleccionados, como lo fue el caso aquí ejemplificado que, ante los hallazgos de una tumoración en el transquirúrgico (no detectada por gabinete convencional), en proceso uncinado de páncreas, corroboró una acertada decisión terapéutica y adecuada técnica quirúrgica.
Sin embargo, la mayoría de estos pacientes mueren de metástasis malignas de TNEP funcionales, sugiriendo que la resección es preferible en una etapa viable para prevenir la invasión metastásica.3-5,8
Para tumores localizados en ámpula de Váter (ampuloma), el tratamiento se basa en el tamaño de la lesión: endoscópico cuando las lesiones son menores a 1 cm; mientras que la escisión transduodenal se emplea en lesiones de entre 1 y 2 cm. Para lesiones mayores a 2 cm está justificado realizar una pancreatoduodenectomía o resecciones segmentarias, aunque los pacientes con lesiones de este tamaño desarrollan tumores recurrentes.12,20
La resección en bloque está indicada en caso de invasión a órganos vecinos (estómago, colon, bazo). En pacientes con resección curativa la supervivencia puede llegar al 72% en 5 años; sin embargo, en aquellos en los que no se puede realizar la resección completa la supervivencia disminuye a un 38%.4,8
A diferencia de lo que antiguamente se creía, la resección quirúrgica de las metástasis hepáticas ofrece ventajas para la supervivencia, y aunque la resección hepática cura menos del 10% de los pacientes afectados que experimentarán una recidiva en los 2 años siguientes a la resección, está indicada en pacientes con metástasis unilobulares que ocupen menos del 75% del parénquima hepático. En caso de una extensión mayor al 75%, el pronóstico es considerado desfavorable y es preferible evitar la cirugía. La resección paliativa está indicada por dolor o síntomas hormonales no controlados cuando pueda extirparse más del 90% del tumor de manera segura.6
Dado lo avanzado de la enfermedad al momento del diagnóstico, muchos pacientes no son candidatos a tratamiento quirúrgico radical; el control del crecimiento tumoral y el control de la actividad hormonal excesiva se logra por medio del tratamiento médico con inhibidores de la secreción, como los análogos de la somatostatina e interferón α. Las formulaciones más eficaces de los análogos de la somatostatina son la octreótida (50, 100 o 500 μg) y el autogel de lanreotida (60, 90 o 120 mg). La dosis inicial de octreótida suele ser de 50 μg por vía subcutánea (SC), dos o tres veces al día; se precisa ajustar el medicamento a la alza. Al cabo de dos semanas se puede cambiar a octreótida LAR (de depósito); estos medicamentos se toleran bien y resultan seguros, con efectos secundarios leves como la diarrea, dolor abdominal, la esteatorrea y la colelitiasis. Dados los efectos antiproliferativos de los análogos de la somatostatina y del interferón α, se ha propuesto que la combinación de ambos fármacos podrían potenciar su acción. Aunque, en un estudio realizado recientemente no hubo ninguna diferencia significativa en las tasas de remisión parcial, enfermedad estable o progresión del tumor entre el grupo de pacientes que tomaban los fármacos como monoterapia y el grupo que los tomaba combinados.21-23
En caso de requerirse coadyuvancia, la utilidad de la quimioterapia citotóxica sistémica aún no está bien definida entre los TNEP carcinoides y gastropancreáticos metastásicos. Entre los preparados utilizados se encuentra la estreptozocina (STZ), el fluorouracilo (FU), la doxorrubicina (DOX) y la clorozotocina/dacarbacina (DTIC), aunque suelen dar tasas bajas de respuesta cuando se emplean como monoterapia. La combinación de estos agentes citotóxicos mejoran la supervivencia. Varios estudios demuestran una respuesta más consistente de los TNEP a la quimioterapia al administrar STZ/DOX, ofreciendo una tasa de regresión tumoral superior a la de STZ/FU (69% frente al 45%), prolongando el tiempo hasta la progresión de la enfermedad (20 frente a 6.9 meses) y también la supervivencia (2.2 frente a 1.4 años).24,25
El gammagrama con octreótide es el método ideal para el seguimiento a largo plazo de estos pacientes, ya que depende de la expresión de receptores de somatostatina en las células tumorales, lo que nos permite la identificación de metástasis y la posibilidad de resección en caso de recidiva, haciendo a este método el más utilizado actualmente.2
El pronóstico de los pacientes con TNEP es difícil de predecir. No es sabido si el pronóstico de los tumores no funcionales es distinto al de los funcionales; a pesar de que algunos estudios han demostrado que la supervivencia de los no funcionales es menor, existen algunos trabajos que no encuentran diferencias significativas. Tampoco se han identificado diferencias consistentes en cuanto a los patrones histológicos entre estos grupos de pacientes, por lo que la definición de malignidad aun hoy en día es ambigua. Algunos autores consideran este término aplicable a un TNEP ante la presencia de metástasis ganglionares o hepáticas, mientras que para otros existen pruebas de malignidad si hay invasión vascular o en estructuras adyacentes.26-28
Podemos concluir que a pesar de tratarse de lesiones infrecuentes, los TNEP deben estar presentes en la mente y en el ejercicio diario del cirujano general ante patología biliar o digestiva, como probabilidad diagnóstica sugerida por una evolución tórpida o un curso agresivo de la enfermedad obstructiva en ausencia de litiasis en la vía biliar (pancreatitis/colangitis de repetición), sustentado con el estudio de gabinete adecuado. Por lo que es importante estar familiarizado con su abordaje diagnóstico y tratamiento actual.
REFERENCIAS
1. Jaramillo-Martínez C, Pantoja-Millán JP. Tumores neuroendocrinos del páncreas. Clin Gastro Mex 2008; 1: 155-78. [ Links ]
2. Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendrocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Onc 2008; 19: 1727-1733. [ Links ]
3. Ramaje JK, Davies AH, Ardill J, Baxt T, Caplin M, Grossman A, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumors. GUT 2005; 54: 1-16. [ Links ]
4. Azimuddin K, Chamberlain RS. The surgical management of pancreatic neuroendocrine tumors. Surg Clin North Am 2001; 81: 511-25. [ Links ]
5. Demeure MJ. Endocrine tumors of the pancreas. En: Clark OH (ed.). Atlas of clinical oncology: Endocrine Tumors. Ontario, B.C: Decker; 2003. p. 177-90 [ Links ]
6. Abood GJ, Go A, Malhotra D, Shoup M. The surgical and systemic management of neuroendocrine tumors of the pancreas. Surg Clin North Am 2009; 89: 249-266. [ Links ]
7. Kazanjian KK, Reber HA, Hines OJ. Resection of pancreatic neuroendocrine tumors: result of 70 cases. Arch Surg 2006; 141: 765-770. [ Links ]
8. Lajous M, Pantoja JP. Tumores neuroendocrinos del páncreas. En: Aguirre DL (ed.). Tratado de cirugía general. México: El Manual Moderno; 2003. p. 1049-60. [ Links ]
9. Perry RR, Vinik AI. Clinical review 72: diagnosis and management of functioning islet cell tumors. J Clin Endocrinol Metab 1995; 80: 2273-2278. [ Links ]
10. Virgolini I, Traub-Weidinger T, Decristoforo C. Nuclear medicine in the detection and management of pancreatic islet-cell tumors. Best Pract Res Clin Endocrinol Metab 2005; 19: 213-227. [ Links ]
11. Rapperport ED, Hansen CP, Kjaer A, Knigge U. Multidetector computed tomography and neuroendocrine pancreaticoduodenal tumors. Acta Radiol 2006; 47: 248-256. [ Links ]
12. Woodside KF, Townsend CM Jr, Evers BM. Current management of gastrointestinal carcinoid tumors. J Gastrointest Sur 2004; 8: 742-756. [ Links ]
13. Sundin A, Eriksson B, Bergstrom M, Langström B, Oberg K, Orlefors H. PET in the diagnosis of neuroendocrine tumors. Ann N Y Acad Sci 2004; 1014: 246-257. [ Links ]
14. Orlefors H, Sundin A, Garske U, Juhlin C, Oberg K, Skogseid B, et al. Whole body (11) C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab 2005; 90: 3392-3400. [ Links ]
15. Eriksson B, Orlefors H, Oberg K, Sundin A, Bergström M, Langström B. Developments in PET for the detection of endocrine tumors. Best Pract Res Clin Endocrinol Metab 2005; 19: 311-324. [ Links ]
16. Rindi G, Capella C, Solcia E. Introduction to a revised clinicopatholgical classification of neuroendcrine tumors of the gastroenteropancreatic tract. Q J Nucl Med 2000; 44: 13-21. [ Links ]
17. Piantino P, Fusaro A, Randone A, Cerchier A, Daziano E. Increased levels of CA19-9, CA 50 and CA 125 in patients with benign disease of the biliary tract and the pancreas. J Nucl Med Allied Sci 1990; 34: 97-102. [ Links ]
18. Pasanen PA, Eskelinen M, Partanen K, Pikkarainen P, Penttilä I, Alhava E. Clinical value of serum tumor markers CEA, CA 50 and CA 242 in the distinction between malignant versus benign diseases causing jaundice and cholestasis; results from a prospective study. Anticancer Res 1992; 12: 1687-1693. [ Links ]
19. Miyakawa S, Ishihara S, Takada T, Miyasaki M, Tsukada K, Nagino N, et al. Flowcharts for the management of biliary tract and ampullary carcinomas. J Hepatobiliary Pancreat Surg 2008; 15: 7-14. [ Links ]
20. Evers BM, Small Bowel. In: Townsend CM Jr (ed.). Sabiston Textbook of Surgery. 16th ed. Philadelphia: WB Saunders; 2001. p. 873-916. [ Links ]
21. Delaunoit T, Neczyporenko F, Rubin J, Erlichman C, Hobday TJ. Medical management of pancreatic neuroendocrine tumors. Am J Gastroenterol 2008; 103: 475-483. [ Links ]
22. Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9: 61-72. [ Links ]
23. Faiss S, Pape UF, Böhmig M, Dörffel Y, Mansmann U, Golder W, et al. Prospective, randomized, multicenter trial on the antiproliferative effect of lantreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors-- the International Lantreotide and Interferon Alfa Study Group. J Clin Oncol 2003; 21: 2689-2696. [ Links ]
24. Moertel C, Lefkopoulo M, Lipsitz S, Hahn RG, Klaasen D. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Eng J Med 1992; 326: 519-523. [ Links ]
25. Kouvaraki MA, Ajani JA, Hoff P, Wolff R, Evans DB, Lozano R, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol 2004; 22: 4762-4771. [ Links ]
26. Hochwald SN, Conlon KC, Brennan MK. Nonfunctional pancreatic islet cell tumors. En: Doherty GM, Skögseid B, editors. Surgical Endocrinology. Philadelphia: Lippincott, Williams and Wilkins; 2001. p. 361-373. [ Links ]
27. White TJ, Edney JA, Thompson JS, Karrer FW, Moor BJ. Is there a prognostic difference between functional and nonfunctional islet cell tumors? Am J Surg 1994; 168: 627-630. [ Links ]
28. Solcia E, Sessa F, Rindi G. Pancreatic endocrine tumors: general concepts, nonfunctioning tumors and tumors with uncommon function. En: Dayal Y, editor. Endocrine pathology of the gut and pancreas. Florida: CRC Press; 1991. p. 105-131. [ Links ]
Nota
Este artículo puede ser consultado en versión completa en: http://www.medigraphic.com/cirujanogeneral

