










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista mexicana de ciencias geológicas
versão On-line ISSN 2007-2902versão impressa ISSN 1026-8774
Rev. mex. cienc. geol vol.26 no.1 Ciudad de México Abr. 2009
Chemical species of chromatite of an industrial landfill in the León valley, Guanajuato, Mexico
Especies químicas de cromatita de un relleno industrial en el valle de León, Guanajuato, México
Lázaro Raymundo Reyes–Gutiérrez1, 2, *, Elizabeth Teresita Romero–Guzmán3, Mildred Guillermina Olmos–Salinas3, 4, and Ramiro Rodríguez–Castillo5
1 División de Geociencias Aplicadas, Instituto Potosino de Investigación Científica y Tecnológica (IPICYT), Camino a la Presa San José 2055, Col. Lomas 4 sección, 78216 San Luis Potosi S.L.P., Mexico. *raymundo.reyes@ipicyt.edu.mx
2 Centro de Investigaciones en Ciencias de la Tierra, Universidad Autónoma del Estado de Hidalgo, Carretera Pachuca–Tulancingo Km. 4.5, Col. Carboneras, 42184, Pachuca de Soto, Hidalgo, Mexico.
3 Departamento de Química, Gerencia de Ciencias Básicas, Instituto Nacional de Investigaciones Nucleares, Carrereta México–Toluca Km. 36.5, 52045, A.P. 18–1027, Mexico.
4 Facultad de Química, Universidad Autónoma del Estado de México, Paseo Tollocan esquina Paseo Colón S/N, 50180 Toluca, Estado de México, Mexico.
5 Departamento de Recursos Naturales, Instituto de Geofísica, Universidad Nacional Autónoma de México, Ciudad Universitaria, 04510 México, D. F., Mexico.
Manuscript received: July 23, 2007
Corrected manuscript received: June 16, 2008
Manuscript accepted: June 22, 2008
ABSTRACT
Chromium is a commonly identified contaminant in soils and groundwater and is widely used in industries. Disposal of industrial solid wastes can cause health and environmental risks due to the leaching and seepage of Cr(VI) from soil to groundwater. In order to improve remediation strategies and make better predictions about the mobility of contaminants, it is critical to understand the time–dependent metal sorption behavior on soil, as well as the mechanism of the sorption reactions, and the dominant chemical species. This study demonstrates that interfacial reactions (e.g., adsorption, desorption, oxidation or reduction) between chromium and minerals play an important role in the spreading of chromium, and could present preferential pathways for chromium mobility in the subsurface environment. Soil samples were collected in a landfill (source) of chromium wastes and their morphology and predominant chemical species were determined. Column experiments were performed on contaminated silty–clayey sand, using deionized water as eluent. It was found in this study that, after 72 h, more than 80% of sorbed chromium was eluted with deionized water, and the total chromium content in the leachate were higher than the permissible limits for human consumption established by Mexican official norms (0.05 mg/L). The Cr(VI) removal efficiency decreased significantly with time and it was independent of the initial pH, indicating that Cr(VI) was poorly adsorbed. UV–Vis analysis indicated that the oxidation state of chromium was Cr(VI). DRX analysis led to the conclusion that CaCrO4 (chromatite), is the main mineral species of Cr(VI), and is predominant in the range of pH from 7 to 8.5. An important effect is that the sorption presented to the pore scale can have consequences on the regional scale because it can retard the mobility of this pollutant during the dispersion process.
Key words: chromium, mobility, contamination, chromatite, Cr(VI), desorption, chemical species, León valley, Mexico.
RESUMEN
El cromo es usado ampliamente en industrias y es un contaminante identificado en suelos y agua subterránea. La disposición de residuos sólidos industriales con niveles de cromo puede provocar riesgos a la salud y al ambiente debido a la lixiviación e infiltración de Cr(VI) hacia el agua subterránea. Para mejorar las estrategias de remediación y entender la movilidad de los contaminantes en el subsuelo, es de suma importancia conocer el comportamiento de la sorción del metal en el suelo con respecto al tiempo de residencia, así como su mecanismo de sorción y la especie química dominante. Este estudio demuestra que las reacciones interfaciales (p.ej., sorción, desorción, oxidación o reducción) entre el cromo y los minerales juegan un papel importante en el transporte del cromo, y puede presentar trayectorias preferenciales para su movilidad en el subsuelo. Se recolectaron muestras de suelo en el interior de un relleno de residuos de Cr(VI) y se determinó su morfología y la especie química predominante. Se realizaron experimentos en columna sobre arena limo–arcillosa contaminada con Cr(VI) empleando agua deionizada como eluyente. En este estudio se encontró que la cantidad de Cr(VI) eluída con agua deionizada fue superior al 80% en un tiempo de 72 h después del contacto con el suelo contaminado, obteniéndose en los luxiviados valores de cromo total mayores al límite permisible para consumo humano establecido en la Norma Oficial Mexicana (0.05 mg/L). La eficiencia de remoción del Cr(VI) disminuye significativamente con el tiempo, es independiente del pH inicial y está en función de la textura del suelo, indicando que el Cr(VI) fue poco adsorbido. Del análisis UV–Vis se concluye que el estado de oxidación del cromo es Cr(VI), el cual predomina en el intervalo de pH de 7 a 8.5. Los resultados por DRX indican que el cromato de calcio (CaCrO4), cromatita, es la principal especie mineralógica de Cr(VI). Un efecto importante es que la sorción que se presenta a la escala de poro puede tener consecuencias en la escala regional en el área de estudio debido a que puede retardar la movilidad de este contaminante en el proceso de dispersión.
Palabras clave: cromo, mobilidad, contaminación; cromatita; Cr(VI), desorción, especies químicas, valle de León, México.
INTRODUCTION
Chromium occurs naturally at trace levels in most soils and water, but disposal of industrial waste and sewage sludge containing chromium compounds has created a number of contaminated sites. Their determination requires the application of a sufficiently sensitive method, but its specificity has an important role to play in the case of chemical individuals or its selectivity in regard to speciation of a group of compounds, e.g., valency states or organic chromium complexes (Swietlik, 1998). Background levels correspond to the total concentrations of metals in soil not affected by human activities. These values have been reported by many authors (e.g., Alloway, 1995; Archer and Hodgson, 1987) and a number of guidelines exist to establish the maximum levels of heavy metals in soils (Council of the European Communities, 1986; U.S. EPA, 1993). Total concentrations of chromium (Cr) in soils range from 9.9 to 121 µg/g (Archer and Hodgson, 1987; ASTM, 2000). Chromium is not present in elemental form and its main source is chromite (FeCr2O4).
There are only a few analytical techniques available that have sufficient sensitivity and selectivity for the direct determination and speciation of trace levels of chromium. Sample pre–treatment techniques, which include analyte element separation and preconcentration, are required in order to determine the low levels of the individual chromium species even when the most sensitive techniques, such as electrothermal atomic absorption spectrometry (ET–AAS), are used. Even though other analytical techniques such as inductively coupled plasma atomic spectrometry (ICP–AS), neutron–activation analysis, electrochemical methods or X–ray fluorescence have been used for this purpose, Atomic Absorption Spectrometry AAS, being a highly selective and element specific technique, is ideally suited for speciation analysis (Sperling et al., 1992).
Chromium has both beneficial and harmful effects on human health (Huffman and Allaway, 1973; Barceló et al., 1993); although Cr(III) is an essential element for human beings and animals, its aqueous concentrations are generally below the US EPA water quality standards of 0.05 mg/L for total chromium because of its low solubility in water. Most of the hexavalent chromium, Cr(VI), compounds are considered toxic and mutagenic because they can produce lung cancer when inhalated and some skin sensibilization problems (Ferguson, 1990; Ewers, 1991; Armienta and Rodríguez, 1995; Barceloux, 1999). In the subsurface, Cr(VI) generally exists in the anionic chromate (CrO42–), (HCrO4–) or dichromate (Cr2O72–) forms, which are relatively soluble and mobile, because they are generally not strongly sorbed by most soils, while the Cr(III) usually occurs in soils as hydroxides that coprecipitate with Fe(III) hydroxides or are adsorbed on soil particles, rarely as oxides, silicates or sulphides (Stollenwerk and Grove, 1985; Kozuh, et al., 2000; Armienta and Quèrè, 2004). Thus, the risk associated with soil and groundwater contamination is high and remediation of soil and groundwater contaminated with Cr(VI) is of critical importance (Armienta and Rodríguez, 1995; Wilkin et al., 2005). It is important to point out that the oxidation state of chromium determines the ecotoxicologic effects on the human. On the other hand, the behavior of chromium in soils depends on various factors, such as the physicochemical properties of soils, the mobility and form of transport of chromium compounds, and the redox reactions which are very important for studying the chromium geochemistry (Palmer and Puls, 1994; Armienta and Quèrè, 2004). The presence of organic matter, Fe(II) and reduced species of sulfur can be responsible for the reduction of Cr(VI) to Cr(III) in natural water, sediments and soils (Kozuh, et al., 2000; Reyes–Gutiérrez and Romero–Guzmán, 2004).
Much research has focused on the remediation of Cr(VI) and several technologies exist for remediation of chromium–contaminated soil and water that fall into five general categories: isolation, immobilization, toxicity reduction, physical separation and extraction. The immobilization approach attempts to reduce the mobility of contaminants by changing the physical or leaching characteristics of the contaminated matrix. Immobilization treatment involves the use of inorganic binders, such as cement, fly ash, lime, slugs, or organic binders, such as bitumen. Among these stabilizing agents, numerous researchers have identified lime, fly ash and Portland cement as the most prominent agents (Catalan et al., 2002; Dermatas and Meng, 2003; Dermatas and Moon, 2006). Many recent experiments have been performed focusing on the stabilizing properties of various iron amendments (Mench et al., 2000; Kim et al., 2003; Warren and Alloway, 2003; Harley et al., 2004). Niu et al. (2005) studied the removal of Cr(VI) from aqueous solutions by means of metallic iron (Fe°) nanoparticles to transform Cr(VI) to nontoxic Cr(III). Different Fe° types were compared under the same conditions. The authors concluded that the Cr(III) compound Cr(OH)3 would be the final product of Cr(VI) reduction, and that iron nanoparticles are a good choice for the remediation of heavy metals in soils and groundwater. Kostarelos et al. (2006) studied the effects of lime and fly ash to immobilize of Cr(VI) in soil. The Cr(VI) immobilization process studied was effective at pH values around 12, with a corresponding overall stabilization degree between 82.0% and 99.8% of the initial chromium content. Laborda et al. (2007) studied the mobility of chromium in compost with no aggressive reagents (ultrapure water with added nitric acid or potassium hydroxide). Kumpiene et al. (2007) evaluated the effects of pH, oxidizing–reducing potential (Eh), liquid–to–solid ratio, presence of organic matter and microbial activity on the mobility of chromium, copper, arsenic and zinc in zerovalent iron (Fe°)–stabilized soil by applying a 25 factorial experimental design. This last study concluded that, at a pH = 3, the release of these elements occur, whereas the Eh measurements seem to have less influence on the remobilization of As and is insignificant for the other elements studied. However, the initially formed nanoparticles tend to either react rapidly with the surrounding media (e.g., dissolved oxygen, sulfate or water) or agglomerate rapidly, resulting in the formation of numerous large particles and rapid loss in reactivity. A new process must be found to prepare physically more stable Fe° nanoparticles.
As part of a global research, this work consider the study case of a thin water–table aquifer associated with sand and gravels, which contains a Cr(VI)–contaminanted plume formed by leachate from an industrial landfill (source) for chromium wastes (Armienta y Rodríguez, 1995). Although chromium contamination in the studied site can originate from a natural source, e.g., in situ weathering of rock minerals leading to metal accumulation in soils (Rodríguez and Armienta, 1995; Robles and Armienta, 2000; Hernández–Silva et al., 2000; Carrillo–Chávez et al., 2003; Vernay et al., 2007), the main contributions are derived from waste disposal on the Química Central (QC) chromate factory lands, which was established in the Buenavista area in 1970 for the production of chromium salts (Armienta and Rodríguez, 1992; Rodríguez and Armienta, 1995; Armienta et al., 1996; Robles and Armienta 2000). During the late 1991, a serious problem of high Cr(VI) concentration in groundwater around QC zone was recognized (Rodríguez et al., 1991).
Aquifers in deposits of unconsolidated sand and gravel are excellent groundwater sources for a variety of uses, but such aquifers are also especially vulnerable to contamination. Groundwater remediation in these aquifers is especially complicated because of the specific lateral and vertical variability of the sedimentological facies (Rodríguez et al., 1991; Reyes–Gutiérrez, 1998; Reyes–Gutiérrez and Romero–Guzmán, 2004). The initial approach to remediation in Buenavista area has involved mass removal of contaminated soils by excavation and of contaminated groundwater by pumping to late 1993. Following initial removal actions, the in situ chemical treatment approach could be undertaken to complete the site and aquifer restoration. Thus, injection of treatment solutions by selected reducing agents (e.g., water, ferrous sulfate, sodium sulfite, sulfurous acid, sodium sulfide, sodium thiosulfate, zerovalent iron, sulfur dioxide, and hydrogen sulfide) upgradient of the groundwater contaminant plume could be undertaken during pumping (Rodríguez et al., 1991; Reyes–Gutiérrez, 1998). Transport of the treatment agent through the contaminated aquifer would serve to immobilize residual contamination and thus to accelerate remediation of the aquifer. Treatment of the unsaturated zone could also be conducted simultaneously with groundwater treatment.
The primary objective of this study is to identify the mechanism of Cr(VI) mobility in the unsaturated zone of the source. In this work, column tests were carried out to desorb Cr(VI) from the soil samples. The specific objectives of this study were (1) to characterize the morphology and mineralogy of soils; (2) to determine the pH and the Cr(VI) concentration of the aqueous fractions obtained from leaching experiments, as well as the total chromium contents in soil samples; (3) to evaluate the residence time of Cr(VI) in the soil; and finally (4) to identify the chemical species in solution, which will determine the dynamic and potential migration of Cr(VI) toward the aquifer.
EXPERIMENTAL
Site description
The study area is located in the northeastern part of the Río Turbio valley, in central–western Guanajuato State, southwest of León city and 7 km away from the city San Francisco del Rincón (Figure 1). The study was conducted inside the facilities of the Química Central (QC) chromate factory, which is located adjacent to the highway and to the railroad León–San Francisco del Rincón, and to the river Los Gómez–León–Turbio and the northwestern edges of San Germán dam. Also, the lands of the QC are located very close to the urban nucleus of Buenavista. The areas situated to the east, northeast and southeast are used for agricultural purposes, mainly for sorghum and lucerne cultivation. The source of Cr(VI) is a landfill for industrial waste 75 m in length, 25 m width and 6 m depth, located inside the QC facilities.
The Cr(VI) leachates derived from the chromium wastes were discharged in a silty sand layer and percolates 3 m towards the water table, initiating the groundwater contamination. The source is located in a shallow aquifer constituted by stratified alluvial material consisting of sand and gravels of variable thickness of approximately 12 to 30 m, with a hydraulic conductivity of 1.27 x 10–3 m/s, and 35% porosity, that rests on a package of clay of variable thickness (Reyes–Gutiérrez, 1998). The top of the sand aquifer consists of a local lens of clay with variable thickness located 6 m under the source (Reyes–Gutiérrez, 1998). The hydraulic gradient in the southwest is about 0.007 and the average groundwater flow velocity ranges from 0.3 to 1.0 m/d (Reyes–Gutiérrez, 1998).
Cr(VI) contamination in the Buenavista area, León, Guanajuato, is associated with a source situated in vadose zone (unsaturated) soils. Leaching of the contaminant from the vadose zone by infiltration processes subsequently results in contamination of the associated shallow aquifer.
Selection of soil samples
Four points were selected for sampling in the QC landfill (Figure 1): an unaltered area of 1 m2 was cleaned and a one meter deep hole was drilled. In the vadose zone, a fine to medium textured sediment was collected. Soil profiles, respectively 30, 60 and 100 cm deep were investigated to assess Cr content. Samples of 2 kg were collected into polyethylene bags, transported in an ice pail to the laboratory and stored in a refrigerator at 4 °C in an in situ moisture condition (0 to ~ 0.1 kPa of water potential). The samples were air dried to retard the activity of Cr(VI) (U.S. EPA, 1996, method 3060A). Besides, a composite sample was prepared by mixing several of the collected samples (100 g), and passing them through a No. 40 mesh to obtain a subsample with a relatively homogeneous particle size; this sample was used in the column experiments.
Leaching experiments
In order to carry out the desorption test, experiments were performed in accordance with the techniques suggested by Cochran (1963) and Reyes–Gutiérrez and Romero–Guzmán (2004). For the leaching experiments, a glass column with 1.5 cm of external diameter and 20 cm in length was packed with 5 g of the composite soil sample. Deionized water was passed through the column, and 10 mL aliquots were collected directly from the column. Seven replicates of the column experiment were carried out and 25 eluted aqueous fractions were collected from each one to determine pH, Cr(VI) and total Cr concentration. In a second experiment, a total of 250 mL of deionized water was eluted through the column for the extraction of Cr(VI); the resulting eluate was air dried and a yellow powder precipitate was obtained. All fractions (contaminated soil, leached soil, precipitate and the leached fractions) were subsequently analyzed.
For the quality control of the experiment, 175 samples of the leached fractions were analyzed and the average of the results was used to control and validate the quality of the Cr(VI) analyses. Although the average value was estimated and used for this purpose, no attempt was made to test the initial data for discordant outliers by using the methodology of Verma and Quiroz–Ruiz (2006a, 2006b). The same was true for other calculations of the average values reported in this paper. The average was reported only if the individual measurements were within an error range of 5% (U.S. EPA, 2000).
Physicochemical characterization
The morphology of the soil particles contaminated with chromium, of the leached soil, and of the chromium precipitates were analyzed in a scanning electron microscope Phillips XL–30 at 25 kV. The samples were mounted on an aluminum holder with a carbon conductive tape, and later covered with a gold layer, approximately 200 Å in thickness, in a Denton Vacuum Desk II sputtering system. Prior to scanning, the fresh prepared soil particles were dried at room temperature for a week. In all cases, the images were taken with a backscattered electron detector. The elemental composition of the same samples was determined by Energy Dispersion Spectroscopy (EDS) with an EDAX DX–4 spectrometer; to obtain the X–ray spectra, a count rate of 2000 to 2500 cps, dead times of 25–30 %, and a 150 seconds acquisition time was used.
A mineralogical analysis by X–Ray Diffraction (XRD) was performed for the soil contaminated with chromium, the leached soil and the chromium precipitates by using a Siemens D–5000 diffractometer with a copper anode X–ray tube. The Kα radiation was selected with a diffracted beam monochromator at 25 kV, and the diffraction pattern was collected from 4 to 70 ° 2θ with a step size of 0.4–0.5° 2θ for 50 minutes in order to acquire X–ray patterns with sufficiently high intensities to identify the minerals present. Compounds were identified by comparing with the Joint Committee on Powder Diffraction Standards (JCPDS; Bayliss, 1986) cards in the conventional way.
The surface area (m2/g) of the soil contaminated with chromium and of the leached soil was determined by the Brunauer, Emmet and Teller (BET) nitrogen adsorption method, with a Micromeritics Gemini 2360. The dry and degassed samples were analyzed using a multipoint adsorption method.
Quantitative analysis of the Cr(VI) fractions recovered from the column experiments were obtained by using UV–Vis Spectroscopy. In order to determine the relationship between UV–Vis absorbance and Cr(VI), solutions of K2Cr2O7 were prepared at various concentration levels in the range of 0 to 100 mg/L from a stock standard solution of chromium (K2Cr2O7, 500 mg/L). All the chemicals used were analytical grade, and solutions were prepared with deionized water. UV–Vis absorption spectra were obtained with a Perkin–Elmer Lambda 25 UV–Vis spectrophotometer at 540 nm by using 1–cm quartz cells. The calibration and the quantification of the recovered fractions were performed in agreement with the Mexican Norm NMX–AA–044–SCFI–2001 (Kozuh, et al., 2000; Xia et al., 2000; Norma Oficial Mexicana, 2001). The method of analysis is based on the redox reaction of 1,5–diphenylcarbazide with Cr(VI) in acidic solution to form the Cr(III)–diphenylcarbazone complex that absorbs electromagnetic energy at 540 nm (Rock et al., 2001). The Cr(VI) solutions obtained were scanned from 200 to 600 nm to identify the chemical species in aqueous solution. The reproducibility (following the method of Santoyo et al., 2006) of the column experiments were tested by making seven replicate measurements on each standard and sample solution. For the quantitative analysis of Cr(VI), a calibration curve (y = 0.03x – 0.0407, r = 0.9982) was used, which was obtained by least–squares regression analysis of a linear plot of the peak area as a function of the standard concentrations. A limit of detection (LOD) of 0.1 mg/L was determined for this method, and the linear dynamic range was from 0 to 100 mg/L with a correlation coefficient (r) of 0.9982 (number of calibration points n=10; statistically significant at two–tailed 99% confidence level; Bevington and Robinson, 2003; Verma, 2005).
The total concentration of chromium in soil samples of the composites used in each experiment was determined with a VARIAN Spectra–10 Plus Atomic Absorption Spectrometer (AAS) adjusted to a wavelength of 357 nm. The detection limit of chromium by this technique is lower than 5 mg/L. The soil samples contaminated with chromium were prepared in accordance with the Mexican Norm NMX–AA–51–1981 (Norma Oficial Mexicana, 1981; Olmos–Salinas, 2006).
RESULTS AND DISCUSSION
The soils contaminated with chromium had adherences of intense yellow color due to the presence of Cr(VI) residues deposited on them. The soil particles are of variable size and heterogeneous form; the color of the raw particles was not distinguished because of the high concentration of chromium deposited on their surface. In the column experiments, the color intensity of the soil disminished as the experiment proceeded, indicating that the deionized water was leaching out the adsorbed chromium from the soil particles.
The morphologic characteristics of the particles of soil contaminated with chromium are showed in Figure 2; a heterogeneous soil is observed, as well as small grumes adhered to particles of major size, indicating that the material is a heterogeneous powder. The size of the discrete soil particles ranges from 1 µm to 200 µm. Besides, elemental chemical analyses of the material, performed with EDS, indicated that the main constituents of the soil are O, Al, and Si, which are the principal components of aluminosilicates (Sposito, 1989), as well as minor contents of elements such as Na, Mg, K, Ca, Cr, and Fe. The averages in weight % of the element concentration in the soil contaminated with chromium are given in the Table 1.
To confirm the removal of the chromium from the soil, a leached soil was analyzed by SEM. Figure 3 shows a SEM micrograph of the leached soil, where the soil particles appear as discrete particles whose size could reach the micron scale. This soil presented a very fine texture, with variable grain size and a light brown color. The EDS analysis demonstrated that the process used for desorbing chromium from the soil was effective, as no chromium was detected in the EDS spectra of this soil sample (Figure 3). The leached soil contains mainly elements such as Si, Al, and O, which are the principal components of the aluminosilicates that form the soil, whereas K, Ca, Fe, Na, and Mg are present in minor amounts. The average concentrations of these elements are presented in Table 1.
The chromium precipitates obtained from the leached fraction are physically a yellow material, similar to that described by Barceloux (1999), formed by thin layers. Figure 4 shows the SEM micrographs of the chromium precipitates; the crystals form hexagonal prisms of variable size. Also, grouped prisms are observed, which grew until they reached a point where the block was deformed. The elemental chemical composition of the desorbed chromium sample consists of Ca and Cr as principal components, as well as O, S, Si, K, and Na as detected by EDS (Table 1).
In agreement with the results, the soil can be described by different entities: aggregates, sheets and inorganic particles that were distributed in the bulk materials. The untreated soil particles are enriched in O, Na, Mg, Al, Si, K, Ti, Ca, Cr and Fe. In addition, the O, Na and Mg contents are lower in the soils with chromium, but the Al, Ca, and Fe contents are lower in the leached soils. The average contents of Si and K found in the contaminated soil remain almost unchanged after leaching.
On the basis of this comparison, it is interpreted that a competition of the chromate anion (CrO42–) sorption sites does not exist for Fe (Reyes–Gutiérrez and Romero–Guzmán, 2004). Finally, the observed higher contents of Ca, Cr and O in the chromium precipitates indicate the presence of a chemical compound constituted by these elements, such as CaCrO4.
The mineralogical analysis by X–ray diffraction (XRD) indicated the formation of chromatite (Figure 5). The diffractogram of the contaminated soil (Figure 5a) shows that the principal minerals found are quartz, SiO2 (JCPDS card 33–1161), albite (Na, Ca)(Si, Al)4O8 (JCPDS card 20–0554), and chromatite, CaCrO4 (JCPDS card 8–0458). In the diffractogram it is possible to estimate that the majority of materials present in the soil with chromium are crystalline.
Figure 5b shows the diffractogram of the leached soil. These samples have a mineralogical composition of albite (Na, Ca)(Si, Al)4O8 (JCPDS card 20–0554), quartz, SiO2 (JCPDS card 33–1161), and calcite CaCO3 (JCPDS card 24–0027). It should be noted that no chromatite was identified by XRD in this sample.
The X–ray diffractogram of the chromium precipitates (Figure 5c) shows that the main crystalline mineralogical phase is chromatite, CaCrO4 (JCPDS card 8–0458), identified by the presence of the 24.5, 33.4 and 49.1° 2θ peaks. This confirm the presence of chromatite, indicated by the Ca, Cr and O contents determined by EDS in the same sample (Table 1). The same chemical species, chromatite, CaCrO4, was reported by Bajda (2005) from soils polluted by electroplating effluents.
Surface area determined by the BET method for the soil contaminated with chromium was of 21.0 ± 0.6 m2 g–1, whereas that of the leached soil was of 70.6 ± 1.8 m2/g. The difference between both soil types (49.6 m2/g) indicates that chromium occupied this surface area.
The 10 mL aqueous fractions obtained from the column experiments showed variable yellow color intensities; color was more intense in the first eluated samples and diminished in subsequent fractions. The monomeric species provide a yellow color to the water when the Cr(VI) concentration is higher than 1 mg/L, however, the water aquires an orange color when it contains high levels of Cr2O72– (Palmer and Puls, 1994). In this study, the yellow color found in the leachates indicates that the chemical element in aqueous solution is Cr(VI).
Figure 6 shows the UV–Vis absorption spectra corresponding to the variable Cr(VI) concentrations of 20 eluted fractions obtained from the column experiments. In each experiment, a total of approximately 25 fractions were eluated, but the high Cr(VI) concentrations in the first five eluted fractions caused the saturation of the absorption bands and are thus not showed in the UV–Vis spectra. In the spectra, two main absorption bands for Cr(VI) are observed, with absorption maxima located at wavelengths of 372 nm and 272 nm, as was reported by Xia et al. (2000).
The chemical analysis of the soluble fraction by UV–Vis spectrometry (Figure 7) allowed to identify Cr(VI) concentrations between 5 and 1,200 mg/L, which are much higher than the permissible limits for total chromium in drinking water established in the Official Mexican Norm (0.05 mg/L; NOM–127–SSA1–1994 – Norma Oficial Mexicana, 1994).
The average Cr(VI) total concentration in the solution was of 2,778 mg/L; this corresponds to the Cr(VI) recovered in the 25 fractions eluated from the column experiments (250 mL) during 27 hours of continuos experiment. When the contaminated soil was saturated with deionized water, the Cr(VI) concentrations in the first recovered fractions were high but rapidly decreased in the subsequent fractions. After 250 ml, Cr(VI) was removed to a concentration of about 5 mg/L (Figure 7). From this plot, it is evident that leaching of Cr(VI) with deionized water did not respond to pH changes and that other mecanism was controlling the sorption process. The observed trend probably suggests that treatment of soil with deionized water results in binding of Cr(VI) through covalent interactions rather than through an ionic bonding, which should be given by ion exchange mechanisms. In this case, a change in pH would not affect the Van der Waals interactions and hence the Cr(VI) behaviour.
Figure 8 shows the distribution chromium species in function of pH in a diagram elaborated for a Cr(VI) concentration of 2,778 mg/L. The diagram shows that above pH = 6.5, the dominant species is CrO42–. As shown in Figure 7, the pH in all studied fractions (7.0 – 8.5; Figure 7) is higher than this threshold value. Besides, though a certain variation in the pH is observed, it is not reflected in changes in the recovered Cr(VI) concentration.
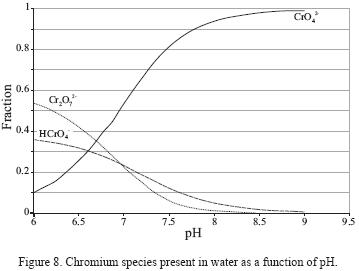
The analyses indicate that deionized water can remove over 79.89% Cr(VI) from soil. The large percentage of Cr(VI) desorbed can indicate the participation of Van der Waals forces, but it can also be related to the formation of a relatively soluble Cr(VI) compound, such as CaCrO4. The Ca2+ cation and CrO42– anion were identified as soluble components, and CaCrO4, chromatite, was identified by X–ray difraction (Figure 5c) in the precipitate recovered from the leachate. Besides, as noted previously, CrO42– is the dominant Cr(VI) species at the pH values found in the solutions.
The mobility of chromium depends on parameters such as the pH, desorption time and volume of deionized water in which the chromium is solvated. The obtained results were plotted in a relative concentration plot (Figure 9) that show the chromium recovery for each aliquot as a function of time. The mobility of Cr(VI) disminishes as the volume of added water increases. It should be noted that up to a final volume of 250 mL, the concentration in the recovered fractions did not fall below the maximum permissible limit of 0.05 mg/L for chromium in water, defined by the Official Mexican Norm (NOM–127–SSA1–1994); the lowest concentration recovered after the addition of 250 mL deionized water was of 2.8 mg/L, and thus for later works it is recommended to use a larger volume of deionized water in order to reach the limit established in the norm.
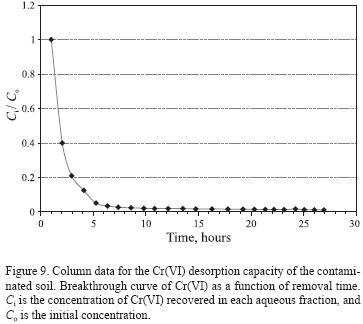
To determine the volume and time at which 50% of the Cr(VI) was separated from the soil, we used breakthrough curves of time versus normalized concentration, Ci /Co, where Ci is the Cr(VI) concentration recovered in each aqueous fraction, and Ci is the initial concentration (Figure 9). It can be observed that the largest amount of Cr(VI) was leached immediately after the beginning of the leaching process, then the Cr(VI) concentration decreased only slightly and reached low values after a short period, indicating that the Cr(VI) removal rates were significantly slower. The time at which 50 % of Cr(VI) was separated was of 2 hours, which indicates that the chromium present in the soil is soluble in deionized water and is liberated very easily; nevertheless, the separation of Cr(VI) after this time is slower. After about 10 hours, the concentration in the solution approached a constant value. Apparently, there was an initial sorption phase which appeared to be complete after 10 hours. With regard to the volume, it follows a similar behavior: the volume at which 50 % of the Cr(VI) was separated from the soil was of approximately 20 mL.
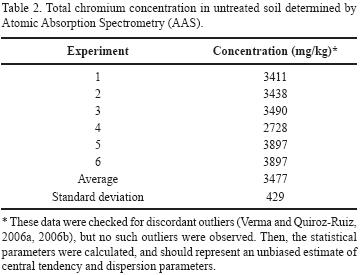
Finally, total chromium concentrations were determined in samples of contaminated soil by AAS, obtaining an average value of 3,477 ± 429 mg/kg (Table 2); the difference between this value and the Cr(VI) analyses correspond to Cr(III). The high total chromium concentration values in the soil are associated with the actual operation of the QC chromate factory. Armienta and Rodríguez (1995) reported a total chromium concentration of 1,000 mg/kg for soil samples collected in the same place at a depth of 30 cm. In 1996, Armienta et al. reported an average content of 2,500 mg/kg total chromium in in the various soil fractions. These data and the new analyses reported in this work indicate that the factory continued increasing the disposal of chromium wastes without treatment. The high solubility of Cr(VI) and the action of rain and irrigation, makes leaching of this species to the soil and deeper subsoil layers possible, resulting in aquifer contamination.
In general, the analysis of the Cr(VI) mobility is relevant because it has an important role in groundwater contamination; for this reason it was necessary to identify its state of oxidation, because the Cr leaching from the vadose zone into the groundwater by infiltration processes is dependent of the oxidation state, as well as other parameters that determine its mobility, such as the coefficient of distribution and retardation (not included in this research study), principally.
This study indicates a high solubility of Cr(VI); this situation can be favorable for the desorption of Cr(VI) through soil washing, under conditions of uniform flow and operating an extraction well (or a set of extraction wells) so that groundwater contaminated with chromatite can be pumped out for subsequent treatment.
CONCLUSIONS
On the basis of the results presented in this study, it can be concluded that the Cr(VI) in the source zone exceeds the maximum permissible limit of 0.05 mg/L (NOM–127–SSA1–1994) for water. But, given its high solubility and mobility in the soil, its mobility in the unsaturated zone can be enhanced by applying the scheme of soil washing so that the contaminant can be removed from the system.
The Cr(VI) was removed in 80 % with deionized water from the soil contaminated with this metal ion. The elemental chemical composition determined by EDS indicate that the contaminated soil contains Ca, K, Na, S, O, Si, Mg, Fe, and Cr, whereas the leached soil did not have measurable amounts of Cr, and contained low weight % of Na, Mg, Al, Si, and Fe. Finally, the precipitate recovered from leachates is mainly composed of Ca, Cr, and O, which indicates the presence of the mineral species chromatite (CaCrO4); this was also verified by XRD.
The minerals identified by XRD in the soil contaminated with chromium were quartz, chromatite, and albite; in the leached soil: calcite, albite, quartz; and in the precipitate recovered from leachates: chromatite.
Desorption of chromium of the surface of sandy soil particles was determined for a pH between a 7.0 and 8.5. It was found that elution with deionized water was efficient and that pH did not affect the removal efficiency. In agreement to the species distribution graph, CrO42– is soluble at this pH, which was corroborated by the recovery of chromatite (CaCrO4 ) precipitates obtained through evaporation of the leachates.
The soil contaminated with chromium in the Buenavista area represents at the moment an important source of hexavalent chromium, that can be leached from the unsaturated zone into the groundwater by infiltration processes. In this work, the chromium mobility in the studied soil was tested only with deionized water, but soil tratment with other chemical reagents should be studied. Moreover, because this study is limited and site specific, the effect of additional components found in natural soils (e.g., organic matter, other clay minerals, etc.) should be considered before clean–up options are evaluated.
ACKNOWLEDGEMENTS
The authors are grateful to Dr. Surendra P. Verma and six anonymous reviewers for the improvement of this paper. We thank Química Central de México for the technical support, to UAEM for financial support through a Project No. 2049/2005 "Evaluación de la capacidad de sorción/desorción del cromo de un relleno industrial", to Q. Leticia Carapia Morales from Instituto Nacional de Investigaciones Nucleares, and to Katie Keller, Ma. Elizabeth Guzmán Keller and Dra. Teresa Orozco for their technical help.
REFERENCES
Alloway, B.J., 1995, Heavy Metals in Soils: London, Glasgow, UK, Blackie and Professional, Second edition, 332 p. [ Links ]
American Society for Testing and Materials (ASTM), 2000, D6232–00, Standard Guide for Selection of Sampling Equipment for Waste and Contaminated Media Data Collection Activities: West Conshohocken, PA, American Society for Testing and Materials, 28 p. [ Links ]
Archer, F.C., Hodgson, I.H., 1987. Total and extractable trace element contents of soils in England and Wales: Journal of Soil Science, 38(3), 421–432. [ Links ]
Armienta, M.A., Quèrè, A., 2004, Hydrogeochemical behavior of chromium in the unsaturated zone and in the aquifer of León Valley, México: Water, Air & Soil Pollution, 84(1–2), 11–29. [ Links ]
Armienta, M.A., Rodríguez, R., 1992, Investigación del impacto ambiental de la dispersión de compuestos de cromo en el área occidental–central del Valle de León, Gto. México: Universidad Nacional Autónoma de México, Instituto de Geofísica, Departamento de Recursos Naturales, Reporte Ténico. [ Links ]
Armienta, M.A., Rodríguez, R., 1995, Environmental Exposure to chromium compounds in the Valley of León, México: Environmental Health Perspectivas, 103(1), 47–51. [ Links ]
Armienta, M.A., Rodríguez, R., Ceniceros, N., Juárez, F., Cruz, O., 1996, Distribution, origin and fate of chromium in soils in Guanajuato, México: Environmental Pollution, 91(2), 391–397. [ Links ]
Bajda, T., 2005, Chromatite CaCrO4 in soil polluted with electroplating effluents (Zabierzow, Poland): Science of the Total Environment, 336(1–3), 269–274. [ Links ]
Barceló, J., Poschenrieder, C., Vázquez, M. D., Gunsé, B., 1993, Beneficial and toxic effects of chromium in plants: solution culture, pot and fields studies, in Vernet, J.P. (ed.), Environmental Contamination: Elsevier, Studies in Environmental Science, 55, pp. 147–169. [ Links ]
Barceloux, D.G., 1999, Chromium: Clinical Toxicology, 37(2), 173–194. [ Links ]
Bayliss, P., 1986, Mineral Powder Diffraction File Date Book: Swarthmore, PA, Joint Committee on Powder Diffraction Standards (JCPDS). [ Links ]
Bevington, P.R., Robinson, D.K., 2003, Data Reduction and Error Analysis for the Physical Sciences: New York, McGraw Hill, 320 p. [ Links ]
Carrillo–Chávez, A., Morton–Bermea, O., González–Partida, E., Rivas–Solórzano, H., Oesler, G., García–Meza, V., Hernández, E., Morales, P., Cienfuegos, E., 2003, Environmental geochemistry of the Guanajuato Mining District, Mexico: Ore Geology Reviews, 23, 277–297. [ Links ]
Catalan, L.J.J., Merlière, E., Chezick, C., 2002, Study of the physical and chemical mechanisms influencing the long–term environmental stability of natrojarosite waste treated by stabilization/solidification: Journal of Hazardous Materials, B94, 63–88. [ Links ]
Cochran, W.G., 1963, Sampling Techniques: New York, USA, John Wiley & Sons, Second edition, 411 p. [ Links ]
Council of the European Communities, 1986, Council directive of 12 June 1986 on the protection of the environment, and in the particular of the soil, when sewage sludge is used in agriculture: Official Journal of the European Communities, L181, 6–12. [ Links ]
Dermatas, D., Meng, X., 2003, Utilization of fly ash for stabilization/solidification of heavy metal containing soils: Engineering Geology 70(3–4), 377–394. [ Links ]
Dermatas, D., Moon, D. H., 2006, Chromium leaching and immobilization in treated soils: Environmental Engineering Science 23(1), 77–87. [ Links ]
Ewers, U., 1991, Standards, Guidelines and Legislative Regulations Concerning Metals and their Compounds, in Merian, E.(ed.) Metals and their Compounds in the Environment: Occurrence, analysis and biological relevance: Weinheim, Verlag Chemie (VCH), 687–711. [ Links ]
Ferguson, J. E., 1990, The Heavy Elements: Chemistry, Environmental Impact and Health Effects: Oxford, Pergamon Press, 211–212. [ Links ]
Harley, W., Edwards, R., Lepp, N.W., 2004, Arsenic and heavy metal mobility in iron oxide–amended contaminated soils as evaluated by short– and long–term leaching tests: Environmental Pollution, 131(3), 495–504. [ Links ]
Hernández–Silva, G., Solorio–Mungía, G., Vasallo–Morales, L., Flores–Delgado, L., Maples–Vermeersch, M, Hernández–Santiago, D., Alcalá–Martínez, R., 2000, Dispersión de Ni y Cr en sedimentos y suelos superficiales derivados de piroxenitas, serpentinitas y basaltos de la cuenca San Juan de Otates, Estado de Guanajuato, México: Revista Mexicana de Ciencias Geológicas, 17(2), 125–136. [ Links ]
Huffman, E.W.D., Allaway, W.H., 1973, Growth of plants in solution culture containing low levels of chromium: Plant Physiology, 52(1), 72–75. [ Links ]
Kim, J–Y., Davis, A.P., Kim, K–W., 2003, Stabilisation of available arsenic in highly contaminated mine tailings using iron: Environmental Science and Technology, 37(1), 189–195. [ Links ]
Kostarelos, K., Reale, D., Dermatas, D., Rao, E., Moon, D.H., 2006, Optimum dose of lime and fly ash for treatment of hexavalent chromium–contaminated soil: Water, Air, and Soil Pollution: Focus, 6(1–2), 171–189. [ Links ]
Kozuh, N., Stupar, J., Gorenc, B., 2000, Reduction and oxidation processes of chromium in soils: Environmental Science and Technology, 34(1), 112–119. [ Links ]
Kumpiene, J., Castillo M.I., Lagerkvist, A.M. , 2007, Evaluation of the critical factors controlling stability of chromium, copper, arsenic and zinc in iron–treated soil: Chemosphere, 67(2), 410–417. [ Links ]
Laborda, F., Górriz, M.P., Bolea, E., Castillo, J.R., 2007, Mobilization and speciation of chromium in compost: A methodological approach: Science of the Total Environment, 373 (1), 383–390. [ Links ]
Mench, M., Vangronsveld, J., Clijsters, H., Lepp, N.W., Edwards, R., 2000, In situ metal immobilisation and phytostabilisation of contaminated soils, in Norman, T., Bañuelos, G. (eds.), Phytoremediation of contaminated soil and Water: Boca Raton, FL, Lewis Publishers, 323–358. [ Links ]
Niu, S.F., Liu, Y., Xu, X.H., Lou, Z.H., 2005, Removal of hexavalente chromium from aqueous solution by iron nanoparticles: Journal of Zhejian University, 6B(10), 1022–1027. [ Links ]
Norma Oficial Mexicana, 1981, NMX–AA–51–1981, Análisis de agua. Determinación de metales. Método Espectrofotométrico de Absorción Atómica: México, Secretaría de Economía. [ Links ]
Norma Oficial Mexicana, 1994, NOM–127–SSA1–1994, Salud ambiental. Agua para uso y consumo humano. Límites permisibles de calidad y tratamientos a que debe someterse el agua para su potabilización, México, Secretaría de Salud, 7 pp. [ Links ]
Norma Oficial Mexicana, 2001, NMX–AA–044–SCFI–2001, Análisis de aguas – Determinación de cromo hexavalente en aguas naturales, potables, residuales y residuales tratadas – Método de prueba: México, Secretaría de Economía, 21 pp. [ Links ]
Palmer, C.D., Puls, R.W., 1994, Natural attenuation of hexavalent chromium in groundwater and soils: United States Environmental Protection Agency, EPA Ground Water Issue, EPA/540/5–94/505, 12 pp. [ Links ]
Olmos–Salinas, M.G., 2006, Remoción de cromo de un relleno industrial: México, Universidad Autónoma del Estado de México, Facultad de Química, Professional thesis,100 p. [ Links ]
Reyes–Gutiérrez, L.R., 1998, Factores que controlan la dispersión de compuestos de cromo en un acuífero de conductividad hidráulica variable: México, Universidad Nacional Autónoma de México, M. Sc. thesis, 150 p. [ Links ]
Reyes–Gutiérrez, L.R., Romero–Guzmán, E. T., 2004, Separación y análisis de un relleno de residuos de cromo en León Guanajuato, in Memorias del XVIII Congreso Nacional de Química Analítica, Pachuca Hidalgo, México, 48 p. [ Links ]
Reyes–Gutiérrez, L.R., Cabral, P. A., Romero–Guzmán, E.T., 2004, Chromium–iron compounds in contaminated soil studied by EDS, XRD and Mossbauer Spectroscopy (abstract), in Lacame 2004, Ninth Latin American Conference on the Applications of the Mössbauer Effect, México, D.F., septiembre 19–24. [ Links ]
Robles, C.J., Armienta, M.A., 2000, Natural chromium contamination of groundwater at León Valley, México: Journal of Geochemical Exploration, 68, 167–181. [ Links ]
Rock, M.L., Bruce, R.J., Helz, G.R., 2001, Hydrogen peroxide effects on chromium oxidation satet and solubility in tour diverse chromium–enriched soils: Environmental Science and Technology, 35(20), 4054–4059. [ Links ]
Rodríguez, R., Armienta, M.A., 1995, Groundwater chromium pollution in the Río Turbio Valley, México: Use of pollutants as chemical tracers: Geofísica Internacional, 34(4), 417–426. [ Links ]
Rodríguez, R., Armienta, M. A., Villanueva S., Díaz P., González T., 1991, Estudio hidrogeoquímico y modelación matemática del acuífero del río Turbio para definir las acciones encaminadas a proteger de contaminantes la fuente de abastecimiento de la Cd. de León Gto.: Universidad Nacional Autónoma de México, Instituto de Geofísica, Comisión Nacional del Agua(CNA)–Secretaría de Agricultura y Recursos Hidráulicos (SARH), Reporte Técnico II, 140 pp. [ Links ]
Santoyo E., Guevara, M., Verma, S.P., 2006, Determination of lanthanides in international geochemical reference materials by reverse–phase high–performance liquid chromatography using error propagation theory to estimate total analysis uncertainties: Journal of Chromatography A, 118(1), 73–81. [ Links ]
Sperling M., Yin X., Welz B., 1992, Differential determination of chromium (VI) and total chromium in natural waters using flow injection on–line separation and preconcentration electrothermal atomic absorption spectrometry: Analyst, 117(3), 629–635. [ Links ]
Sposito, G., 1989, The Chemical of Soils: New York, Oxford University Press. 304 p. [ Links ]
Stollenwerk, K.G., Grove, D.B., 1985, Adsorption and desorption of hexavalente chromium in an alluvial aquifer near Telluride, Colorado: Journal Environmental Quality, 14(1), 150–155. [ Links ]
Swietlik, R., 1998, Speciation Analysis of Chromium in Waters: Polish Journal of Environmental Studies, 7(5), 257–266. [ Links ]
United States Environmental Protection Agency (U.S. EPA), 1993, Standards for the use or disposal of sewage sludge; Final rules: United States Environmental Protection Agency, 40 CFR Parts 257, 403 and 503, Federal Register 58, 9248–9415. [ Links ]
United States Environmental Protection Agency (U.S. EPA), 1996, Soil Screening Guidance: User's Guide: Washington, DC, United States Environmental Protection Agency Office of Solid Waste and Emergency Response, EPA/540/R–96/0180, 178 p. [ Links ]
United States Environmental Protection Agency (U.S. EPA), 2000, Guidance for Data Quality Assessment: Practical Methods for Data Analysis (QA/G–9): Washington, DC, United States Environmental Protection Agency, Office of Research and Development, EPA/600/R–96/084, 219 p. [ Links ]
Verma, S.P., 2005, Estadística Básica para el Manejo de Datos Experimentales; Aplicación en la Geoquímica (Geoquimiometría): México, D.F., Universidad Nacional Autónoma de México, 186 p. [ Links ]
Verma, S.P., Quiroz–Ruiz, A., 2006a, Critical values for 22 discordancy test variants for outliers in normal samples up to sizes 100, and applications in science and engineering: Revista Mexicana de Ciencias Geológicas, 23(3), 302–319. [ Links ]
Verma, S.P., Quiroz–Ruiz, A., 2006b, Critical values for six Dixon tests for outliers in normal samples up to sizes 100, and applications in science and engineering: Revista Mexicana de Ciencias Geológicas, 23(2), 133–161. [ Links ]
Vernay, P., Gauthier–Moussard, C., Hitmi A., 2007, Interaction of bioaccumulation of heavy metal chromium with water realtion, mineral nutrition and photosynthesis in developed leaves of Lolium perenne L.: Chemosphere, 68(8), 1563–1575. [ Links ]
Warren, G.P., Alloway, B.J., 2003, Reduction of arsenic uptake by lettuce with ferrous sulphate applied to contaminated soil: Journal of Environmental Quality, 32, 767–772. [ Links ]
Wilkin R.T., Su C., Ford R.G., Paul C.J., 2005, Chromium removal processes during groundwater remediation by a zerovalent iron permeable reactive barrier: Environmental Science and Technology, 39(12), 4599–4605. [ Links ]
Xia, L., Akiyama, E., Frankel, G., McCreery, R., 2000, Storage and release of soluble hexavalente chromium from chromate conversion coatings. Equilibrium aspects of Cr(VI) concentration: Journal of the Electrochemical Society, 147(7), 2556–2562. [ Links ]

