










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.4 Ciudad de México oct./dic. 2004
Revisión
Pincer Complexes. Applications in Catalysis
David Morales-Morales*
Instituto de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, Circuito Exterior, Coyoacán, 04510 México D. F. Tel. +52-55-56224514, Fax +52-55-56162217. E-mail: damor@servidor.unam.mx
Recibido el 18 de octubre del 2004.
Aceptado el 7 de diciembre del 2004.
Abstract
In the recent years the development of complexes having in its structure pincer type ligands has received considerable attention. Now a days these complexes are designed and synthesized not just for the mere structural interest, but for the great number of potential applications that these compounds may have, including their use as robust catalysts, motifs in molecules that exhibit self assembly arrangements, synthons for their employment in Medicinal Chemistry and as fundamental components in the synthesis of dendrimeric materials for their potential use in catalysis. Thus, the present review shows the multiple applications that the pincer complexes have had in particular in the area of catalysis and the future perspectives of applications of these compounds in chemistry.
Key words: PCP pincer ligands, SCS pincer ligands, NCN pincer ligands, pincer type complexes, catalysis.
Resumen
En los últimos años el desarrollo de la Química de compuestos conteniendo ligantes tipo pincer (pinza para su traducción en español) ha tenido un gran avance, pasando de ser especies sintetizadas meramente por su interés estructural a formar parte hoy en día de uno de los motivos en Química más utilizados por los químicos para el desarrollo de especies con diversas aplicaciones, que van desde su uso como precursores de catalizador, moléculas de interés en procesos de autoensamblado, sintones empleados en Química medicinal y el uso de estas moléculas como componentes fundamentales en la síntesis de materiales dendriméricos para usos múltiples entre los cuales resalta su profuso empleo en catálisis. De esta forma, la presente revisión representa una compilación de los múltiples y variados casos en los que compuestos tipo pincer han sido empleados en procesos catalíticos así como las perspectivas de desarrollo de esta área.
Palabras clave: Ligandos tipo pinza PCP, Ligandos tipo pinza SCS, Ligandos tipo pinza NCN, complejos con ligantes tipo pinza, catálisis.
Dedicated to the memory of Dr. Raymundo Cruz Almanza
Introduction
In 1976 Moulton and Shaw [1] synthesized for the first time a pincer type ligand. In that point this ligand and its corresponding complexes only represented some very novel derivatives of a new diphosphine. These ligands and their complexes were forgotten for one decade. Then in the 80's a careful reexamination of the properties of these complexes revealed this compounds to have an extraordinary thermal stability, given their high melting points (they sublime without decomposition), property that could enable these complexes to be potentially used in homogeneous catalysis. Now a days these species have been motif of multiple studies beyond catalysis, ranging from application in nanoscience to the development of chemical sensors and chemical switches [2].
The pincer type complexes consist on a metallic center and of course a pincer type ligand containing in its structure the donor atoms being thus capable of coordinate in a tridentated manner to acquire the typical arrangement of the pincer like ligands.
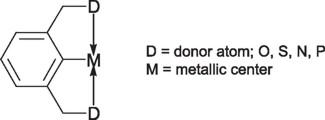
It is believed that is the σ metal-carbon bond the responsible for the unique stability of these complexes, thus avoiding the dissociation of the metal from the ligand and thus the decomposition of the complex, while the donor atoms and their corresponding substituents allow the fine tuning of the steric and electronic properties. Moreover, modification of these substituents has allowed in the recent years to incorporate stereochemical centers capable of induce chirality during a given process. Thus, depending of the donor atoms in the pincer ligands, these compounds are denominated as PCP pincer complexes for those compounds having phosphorus in their structures, SCS for those containing sulfur, etc.
The facility to modify and tune the properties of these ligands and their complexes has been reflected in the profuse employment of these species in different areas of chemistry, particularly in catalysis [3].
Heck Reaction
Since its discovery in the 60's, the Heck reaction has turned into a real power tool in organic synthesis, now a day reaching the status of angular stone in the modern organic synthesis [4]. In general the Heck reaction consists in the coupling of an α-olefin with a bromo or iodo derivative. Most of the processes involving the Heck reaction are catalyzed by Pd(II) or Pd(0) derivatives in the presence of PPh3 in excess.
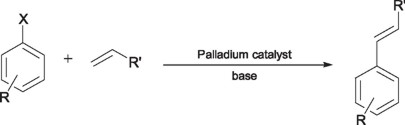
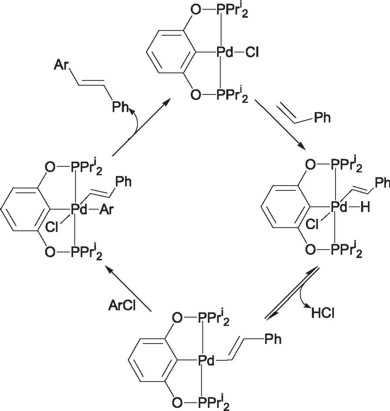
Unfortunately, the reaction intermediates formed during the catalytic reaction are sensitive to oxygen or thermally unstable, hampering the coupling process. In the recent years several research groups have done important advances with the aim of getting the ideal catalyst with the proper characteristics of reactivity and stability to carry out this process, the result of these experiments has lead to the researchers to the use of ortho metallated complexes, among which the pincer type ligands represent one of the most important examples. Milstein and coworkers were the first to employ Pd(II)-PCP pincer complexes (1, 2) in the Heck coupling reaction.
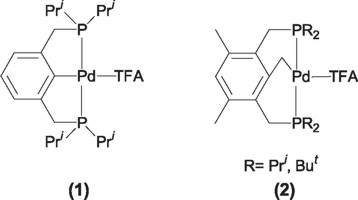
Milstein found that these complexes were active without decomposition at reaction temperatures as high as 140 °C, over reaction periods of 300 hours or higher. By using these catalysts (1, 2) Milstein achieved full conversion in the couplings of iodobenzene with methylacrylate, using N-methylpyrrolidine (NMP) as solvent and sodium carbonate as base with a maximum of 500,000 turnover numbers for iodobenzene and up to 132,900 for bromobencene.
By the same time, Beller and Zapf [6] reported the use of electro-attractor phosphite ligands for the Heck couplings of activated chlorobenzenes.
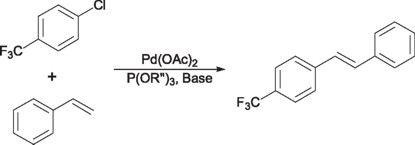
Based on these results Jensen [7] et. al synthesized an analogous PCP pincer type ligand based on phosphinito fragments as P donors. The palladium derivatives of this ligand (3) where shown to be efficient in the coupling of chlorobenzenes, being one of the few examples then known to activate, deactivated or sterically hindered chlorobenzenes.
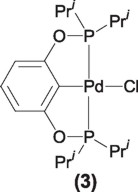
This complex (3) showed to be as reactive as the PCP phosphino derivative reported previously by Milstein.
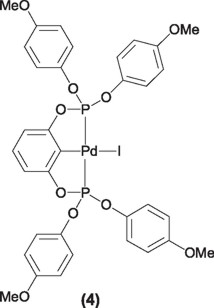
A similar approach lead to Shibasaki [8] et. al, to the synthesis of complex (4), this compound was able to provide turnover numbers higher that 980,000.
Several other pincer type complexes and their palladium derivatives have been synthesized and used in the Heck reaction, including CNC ligands where C represents a carbon from a heterocyclic carbene [9] (5, 6) and SCS ligands (7) [10].
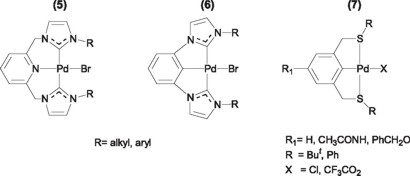
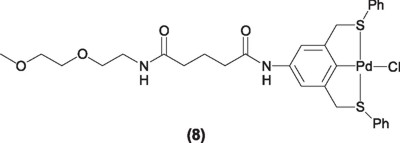
These complexes have demonstrated analogous reactivities and stabilities as their phosphinated counterparts. However, an interesting characteristic of the SCS pincer ligands is the facile functionalization of the aromatic backbone to attach tails that in turn can be linked to polyethylene glycol supports (8), thus generating highly stable and highly active catalyst that can be easily separated, isolated and then used again for at least three cycles without decrease in activity [10].
Thus, characteristics like the supreme activity and superior thermal stability that the palladium pincer complexes have shown in the C-C coupling reactions (Heck type reactions), has lead to Jensen and Morales-Morales [7] to propose an alternative reaction mechanism involving Pd(II)/Pd(IV) [5,7] species instead of the traditionally accepted mechanism involving Pd(0)/Pd(II) species. This reaction mechanism is still under debate.
Suzuki-Miyaura Couplings
The Suzuki or Suzuki-Miyaura C-C couplings [11] consist in the reaction of a halobenzene with arylboronic acids in the presence of a base. This reaction proceeds by a similar reaction mechanism as that of the Heck reaction, thus most of the catalysts usually employed in the Heck coupling reactions have been successfully employed in the Suzuki reaction too.
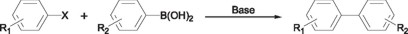
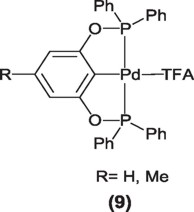
Thus, Pd(II) complexes having phosphinito PCP pincer ligands (9) have been successfully used by Bedford et. al [12] in the couplings of aryl halides with phenyl boronic acid, exhibiting quantitative yields and turnover numbers in the order of 92,000. These complexes are also efficient in the couplings of deactivated and sterically hindered aryl bromides.
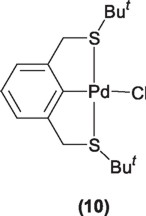
On the other hand, Pd(II) SCS pincer complexes (10) have also been employed in the Suzuki type couplings, however their application has been limited being only active in the reaction of p-bromotoluene with phenyl boronic acid to a maximum of 69% yield of the corresponding biphenyl [13].
Dehydrogenation of alkanes
Saturated hydrocarbons represent one of the most abundant and accessible feedstocks in our planet [14]. However, their use has been limited due to the intrinsic lack of reactivity of these compounds [15]. An interesting alternative to this problem has been the use of transition metal complexes able to activate C-H bonds under mild reaction conditions [16]. Some of these complexes have shown activity in the dehydrogenation of alkanes to alkenes. However, the extremely low reaction rates and the low turnover numbers or the instability of the employed catalysts under the reaction conditions16 has limited the use of these species.
In 1976 Moulton and Shaw reported [1] for the first time the synthesis of the complex IrHCl{C6H3-2,6-(CH2PBut2)2} (11). They observed that this compound exhibited a high thermal stability, subliming without visible decomposition at temperatures as high as 180 °C. These results, led independently to the research groups of Jensen [16], Goldman [18] and Leitner [19] to the use of derivatives of this complex for its potential application as catalysts in the dehydrogenation of alkanes. Thus in 1998, Jensen and coworkers [20] reported the use of the dihydride rhodium complex RhH2{C6H3-2,6-(CH2PBut2)2}(12) in the dehydrogenation of cyclooctane at 150 °C, using tert-butylethylene as sacrificial hydrogen acceptor. This compound has been tested at reaction temperatures as high as 200 °C, showing to be stable for periods of weeks. However the dehydrogenation reaction using this complex only afforded 1.8 turnovers at 200 °C. Further experiments, led Jensen and coworkers [21] to determine that it was in fact the iridium derivative IrH2{C6H3-2,6-(CH2PBut2)2} (13) and not the rhodium complex (12) the best dehydrogenation catalyst. Thus, under the optimized conditions Jensen and coworkers were able to attain turnover numbers of 720 at reaction temperatures of 200 °C, even though this complex exhibits excellent activity at temperatures as low as 150 °C (TON = 82). Unfortunately, complex (13) is quickly deactivated by the formed product (product inhibition) [21].
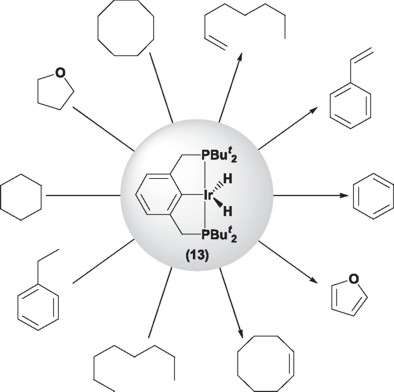
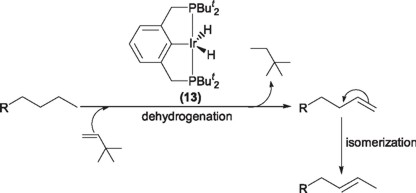
The complex IrH2{C6H3-2,6-(CH2PBut2)2} (13), has been employed in the activation of C-H bonds of several substrates (vide supra) [21]; probably the most notable case being the dehydrogenation of linear alkanes to their corresponding terminal alkenes (α-olefins), being this the kinetically favored process. However, the same complex slowly catalyzes the isomerization of the terminal alkene to internal alkenes, being this the thermodynamic product [22].
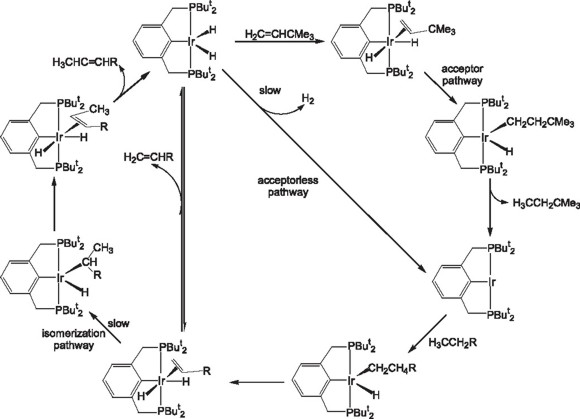
Further modifications in the PCP pincer ligand, like changing substituents at the P moiety and different hydrogen acceptors has led to Goldman and coworkers [23] to increase the efficiency of the catalytic system to a maximum of 68% selectivity for the terminal alkene in the catalytic dehydrogenation of n-octane with turnover numbers in the order of 143. Further improvement to the system has been the elimination of the need of the sacrificial hydrogen acceptor, this achievement being the product of the join efforts of the Jensen and Goldman research groups, reaching a 1000 turnovers under the optimized acceptorless conditions [24]. Further contribution by theoretical calculations [25] have led to the postulation of a tentative reaction mechanism through which complexes IrH2{C6H3-2,6-(CH2PBut2)2} (13) and IrH2{C6H3-2,6-(CH2PPri2)2} (14) carried out the alkane dehydrogenation both under hydrogen acceptor and acceptorless conditions [26].
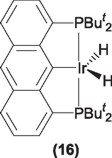
Recently, Kaska and coworkers [27] have reported a rigid PCP pincer system based on antracene backbone (16), the iridium derivative of this ligand has demonstrated catalytic activity in the dehydrogenation of alkanes at temperatures as high as 250 °C without decomposition. Although even at this reaction temperature the system do not match the performance of the complexes 13 and 14, nor in yield of the terminal olefin or in the turnover numbers previously discussed.
Hydrogen Transfer Reactions
van Koten and coworkers have employed successfully ruthenium NCN (17) and PCP (18) pincer complexes to perform hydrogen transfer reactions to reduce ketones to their corresponding alcohols using iso-propanol as source of hydrogen and KOH as cocatalyst [28].
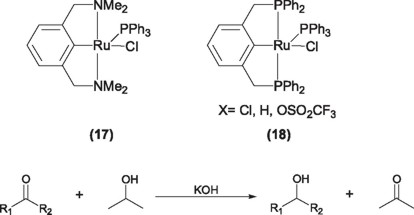
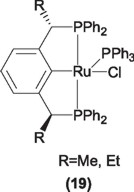
Although both complexes are active catalysts in this process, the best yields and turnover numbers where obtained with the PCP pincer derivatives, for instance, for cyclohexanone, under reflux conditions, yields of 98% and turnover numbers of 27,000 were obtained, being these numbers the best so far obtained, compared with mono phosphine ruthenium complexes like [RuCl2(PPh3)] or [RuCl(H)(PPh3)] [29]. Given these results, several attempts have been done with the aim of interpolate this process to obtain enantiomerically pure alcohols, among these the use of chiral ruthenium PCP pincer complexes (19) [30]. However, despite of the fact that complex 19 exhibits catalytic activity comparable to the non-chiral counterparts, the reaction of iso-PrOH and acetophenone only affords 14% ee [31].
Aldolic Condensations
The synthesis of enantiomerically pure oxazolines can be conveniently done through the gold catalyzed [32] aldolic condensation reactions between an aldehyde or a ketone with an isocyanate. These compounds are very important, since the consecutive hydrolysis of the oxazolines obtained by this procedure, offers a simple and efficient route for the synthesis of β-hydroxyaminoacids.
It is noteworthy that the first enantiomerically pure PCP pincer complexes were evaluated in this process. Venanzi and coworkers [33] designed these compounds by introducing chiral acetals in the benzylic positions of the PCP ligands, the reaction being achieved by the Sharpless type enantioselective epoxydation.
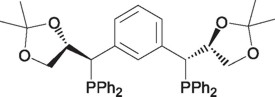
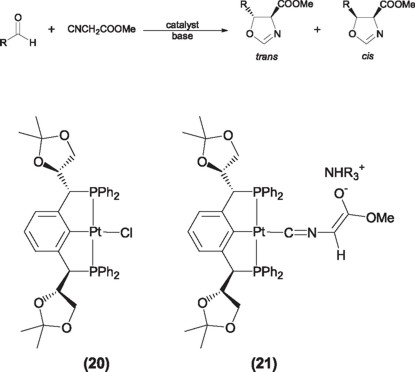
The platinum derivative of this ligand (20) has shown to be active in the asymmetric aldolic addition of methyl-α-isocyanoacetate with aldehydes to yield moderated enantiomeric yields of 32% and 65% ee for the corresponding cis and trans oxazolines respectively. This reaction is carried out in the presence of a base that acts as cocatalyst for the formation of the metal-isocyanoenolate intermediate (21).
However, the synthesis of these compounds is difficult and tedious, thus its potential application has been limited. Recently, Zhang and coworkers [34] have reported an easier and efficient route for the synthesis of enantiomerically pure PCP pincer ligands and their Pd(II) complexes (22).
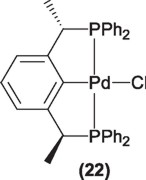
The use of the palladium derivatives of this ligand (22) in the same reaction reported by Venanzi, affords the formation of oxazolines in high yields, with high enantioselectivity for the formation of the cis-oxazolines. This example once more illustrates the importance of the proper selection of the ligand and metal for a particular transformation.
Other chiral pincer type complexes have also been employed. For instance, ligands including ozaxolines in their backbones afford modest results in the synthesis of oxazolines [35].
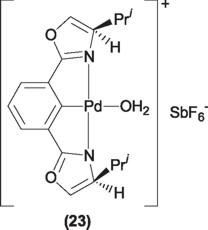
In addition, SCS pincer ligands including chiral groups have also been employed in this reaction. Unfortunately, the chiral induction using this kind of complexes resulted to be zero, this being probably due to the fact that the chiral centers are located too far from the metal center to have a significant effect in the chiral discrimination during the reaction [36].
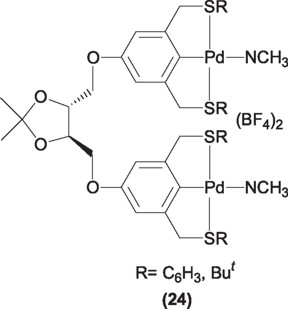
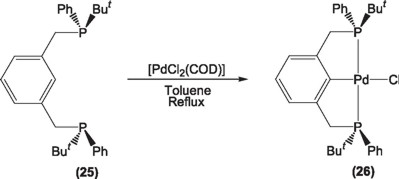
Most recently Morales-Morales and coworkers [37] have synthesized a PCP pincer ligand (25) and its palladium derivatives (26) having the chiral centers at the phosphorus atoms and used them in the same reaction with poor enantiomeric excesses in the order of 6%. In this case it does seems that the R groups at the P* chiral centers are too similar in size to led to a good chiral discrimination, thus efforts pointing to the design of similar ligands with bulkier requirements are under way.
Asymmetric allylic alkylation
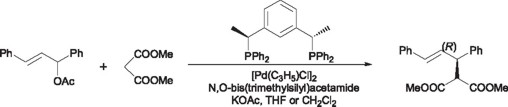
In the recent years, the allylic alkylation reaction in its asymmetric fashion has received considerable interest; proof of this is the tremendous number (more than a 1000) of chiral diphosphines that have been synthesized for this particular reaction. In the field of the pincer ligands Zhang and coworkers [38] have used the PCP pincer chiral ligand 22, previously employed in the aldolic condensation reaction, in the reaction of dimethyl malonate with 1,3-diphenyl-2-propenyl acetate using [Pd(C3H5)Cl2]2 as a source of palladium, attaining yields in the order of 50 to 94%. This particular reaction proceeds at room temperature; however decreasing of the reaction temperature increases considerably the enantiomeric yields to a maximum of 79% ee for the enantiomer (R).
Recent advances
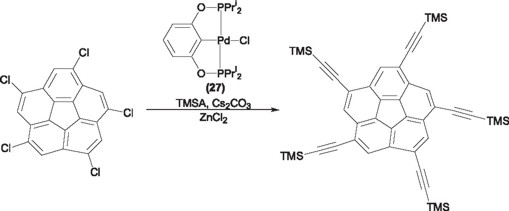
As it can be noted from the above, the pincer ligands an their metallic derivatives represent in many cases the ideal examples to carry out catalytic processes otherwise difficult or impossible to be carried out with conventional diphosphine ligands. The versatility of these species to be modified and modulated both steric and electronically, makes these compounds an their corresponding transition metal derivatives attractive complexes to be continuously used in different challenging catalytic processes. Thus, around the world several important research groups maintain as priority research lines the design of new pincer ligands, their complexes and their potential applications, while other research groups have foreseen the potential of these complexes by use them as catalyst in different organic transformations. It is noteworthy the use of the palladium-phosphinito PCP pincer complex 27 [39] in the synthesis of corannulenes in high yields recently reported by Siegel and coworkers [40].
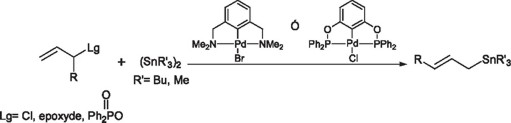
Moreover, Zsabó and coworkers [41] have found the palladium derivatives of NCN and PCP pincer complexes to be active catalysts in the allylic stannylation reaction with high yields.
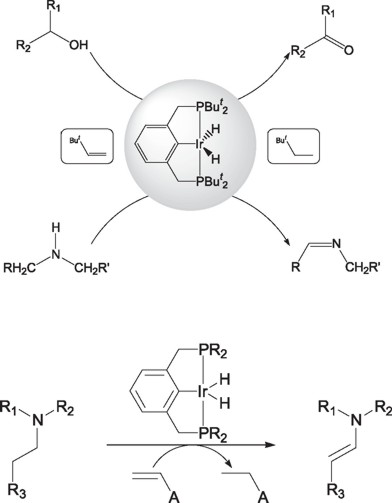
Recently, Morales-Morales and coworkers have extended the application of the iridium pincer complexes IrH2{C6H3-2,6-(CH2PR2)2} (R= But, Pri) for their application in the catalytic dehydrogenation of amine [42] and alcohols [43], providing alternative high yield methods for the synthesis of imines and ketones and aldehydes.
Additionally, Goldman et. al [44] has recently reported the use of these same species for the catalytic dehydrogenation of tertiary amines to enamines in good yields.
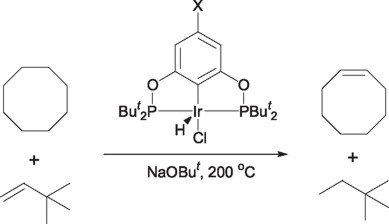
Furthermore, Brookhart et al. [45] and Morales-Morales et al [46] have recently reported, independently, the use of Iridium-Phosphinito PCP pincer type complexes in the catalytic dehydrogenation of alkanes, thus matching the yields and performance of the previously reported phosphino analogues. However, these complexes exhibit additional advantages compared to their phosphino counterparts, such as the fact that the phosphinito PCP pincer ligands can be obtained in an easier manner and in higher yields; and, second, that the hydrido species are not necessary, since this complex can be formed in situ by plain addition of a strong base, like NaOBut.
The continuous search for more specific ligands [47], has taken several research groups to the synthesis of more complex ligands as well as their transition metal complexes, with more elaborated structures including fullerene (28) [48] or ferrocene (29) [49] backbones or very sterically hindered ligands (30) that have allowed to block, in a very specific way, particular quadrants in the complex and thus make them able to provide exceedingly good enantiomeric excess values in, for instance, asymmetric Michael additions [50].
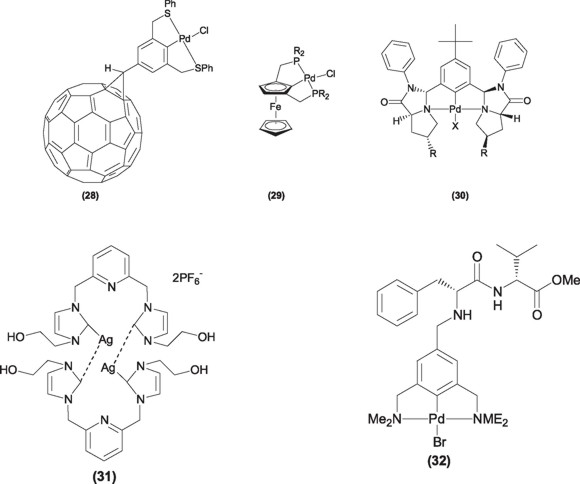
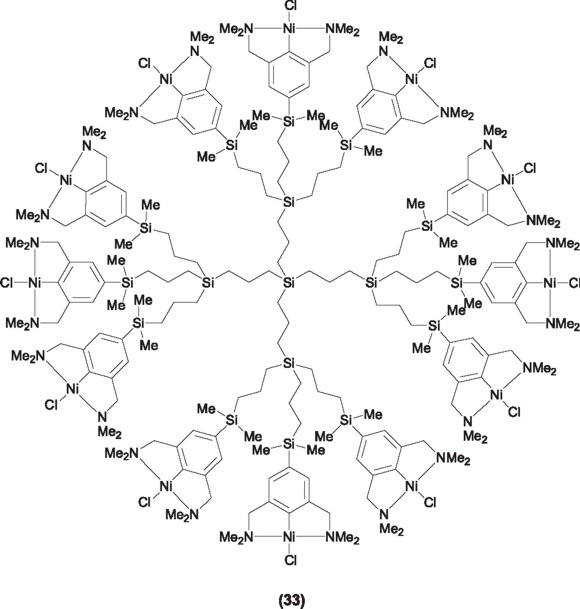
It results easy to forecast that the chemistry of pincer ligands would continue to grow and evolve in the following years, to be included in new and emerging areas of chemistry. Now a days silver(I) systems having the pincer motif in their structure have been considered for their application in Medicinal Chemistry as antimicrobial agents (31) [51], bioorganometallic chemistry (32) [52], and more recently used in the design of nanostructured and dendrimeric systems for their application in catalytic heterogeneized systems and supramolecular chemistry (33) [53].
References
1. Moulton, C. J.; Shaw, B. L. J. Chem. Soc., Dalton Trans. 1976, 1020-1024. [ Links ]
2. van Koten, G.; Albrecht, M. Angew. Chem. Int. Ed. 2001, 40, 3750-3781. [ Links ]
3. van der Boom, M. E.; Misltein, D. Chem. Rev. 2003, 103, 1759-1792. [ Links ]
4. Heck, R. F. J. Am. Chem. Soc. 1968, 90, 5518-5526. [ Links ] b) Heck, R. F. Org. React. 1982, 27, 345-390. [ Links ] c) Heck, R. F. Comprehensive Organic Synthesis: Selectivity, Strategy and Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I., Eds.; 1991; Vol. 4. p 833, Chapter 4.3. [ Links ] d) Cabri, W. Acc. Chem. Res. 1995, 28, 2-27. [ Links ] e) Beletskaya, I. P.; Cheprakov, A.V. Chem. Rev. 2000, 100, 3009-3066. [ Links ]
5. Ohff, M.; Ohff, A.; van der Boom, M. E.; Milstein, D. J. Am. Chem. Soc. 1997, 119, 11687-11688. [ Links ]
6. Beller, M.; Zapf, A. Synlett 1998, 792-794. [ Links ]
7. Morales-Morales, D.; Redón, R.; Yung, C.; Jensen, C. M. Chem. Commun. 2000, 1619-1620. [ Links ] b) Morales-Morales, D.; Grause, C.; Kasaoka, K.; Redón, R.Cramer, R. E.; Jensen, C. M. Inorg. Chim. Acta 2000, 300-302, 958-963. [ Links ]
8. Miyazaki, F.; Yamaguchi, K.; Shibasaki, M. Tetrahedron Lett. 1999, 40, 7379-7383. [ Links ]
9. Peris, E.; Loch, J. A.; Mata, J.; Crabtree, R. H. Chem. Commun. 2001, 201-202. [ Links ] b) Grundemann, S.; Albrecht, M.; Loch, J. A.; Faller, J. W.; Crabtree, R. H. Organometallics 2001, 20, 5485-5488. [ Links ] c) Tulloch, A. A. D.; Danopoulos, A. A.; Tizzard, G. J.; Coles, S. J.; Hursthouse, M. B.; Hay-Motherwell, R. S.; Motherwell, W. B. Chem. Commun. 2001, 1270-1271. [ Links ]
10. Arroyo, M.; Cervantes, R.; Gómez-Benítez, V.; López, P.; Toscano, R.A.; Morales-Morales, D.; Torrens, H. Synthesis-Stuttgart. 2003, 1565-1568. [ Links ] b) Bergbreiter, D. E.; Osburn, P. L.; Liu, Y.-S. J. Am. Chem. Soc. 1999, 121, 9531-9538. [ Links ]
11. Suzuki, A. J. Organomet. Chem. 1999, 576, 147-168. [ Links ] b) Suzuki, A. Chem. Rev. 1995, 95, 2457-2483. [ Links ]
12. Bedford, R. B.; Draper, S. M.; Scully, P. N.; Welch, S. L. New J. Chem. 2000, 24, 745-747. [ Links ]
13. Zim, D.; Gruber, A. S.; Ebeling, G.; Dupont, J.; Monteiro, A. L. Org. Lett. 2000, 2, 2881-2884. [ Links ]
14. Jones, D. W. Science 2000, 287, 1942. [ Links ]
15. Kakiuchi, F. and Murai, S. In Activation of Unreactive Bonds and Organic Synthesis, S. Murai, Ed. Springer-Verlag, Berlin, 1999, chap. 3, pp. 56-58. [ Links ]
16. Crabtree, R. H.; Mihelcic, J. M.; Quirk, J. M. J. Am. Chem. Soc. 1979, 101, 7738; [ Links ] b) Baudry, D.; Ephritikhine, M.; Felkin, H.; Holmes-Smith, R. J. Chem. Soc. Chem. Commun. 1983, 788; [ Links ] c) Felkin, H.; Fillebeen-Khan, T.; Holmes-Smith, R.; Zakrzewski, J. Tetrahedron Lett. 1964, 25, 1279; [ Links ] d) Felkin, H.; Fillebeen-Khan, T.; Holmes-Smith, R. Lin, Y. Tetrahedron Lett. 1985, 26, 1999; [ Links ] e) Burk, M. W.; Crabtree, R. H.; McGrath, D. V. J. Chem Soc. Chem. Commun. 1985, 1829; [ Links ] f) Burk, M. W.; Crabtree, R. H. J. Am. Chem. Soc. 1987, 109, 8025. [ Links ]
17. Gupta, M.; Hagen, C.; Flesher, R. J.; Kaska, W. C.; Jensen, C. M. Chem. Commun. 1996, 2083-2084. [ Links ]
18. Wang, K.; Goldman, M. E.; Emge, T. J.; Goldman, A. S. J. Organomet. Chem. 1996, 518, 55-68. [ Links ]
19. Leitner, W.; Six, C. Chem. Ber./Recueil. 1997, 130, 555-558. [ Links ]
20. Lee, D. W.; Kaska, W. C.; Jensen, C. M. Organometallics 1998, 17, 1-3. [ Links ]
21. Gupta, M.; Hagen, C.; Kaska, W. C.; Crammer, R. E.; Jensen, C. M. J. Am. Chem. Soc. 1997, 119, 840-841. [ Links ] b)Gupta, M.; Kaska, W. C.; Jensen, C. M. Chem. Commun. 1997, 461-462. [ Links ] c) Gómez-Benítez, V.; Redón, R.; Morales-Morales, D. Rev. Soc. Quim. Mex. 2003 (47) 124-126. [ Links ]
22. Liu, F.; Pak, E. B.; Singh, B.; Jensen, C. M.; Goldman, A. S. J. Am. Chem. Soc. 1999, 121, 4086-4087. [ Links ]
23. Liu, F.; Goldman, A. S. Chem. Commun. 1999, 655-656. [ Links ]
24 Xu, W.; Rosini, G. P.; Gupta, M.; Jensen, C. M.; Kaska, W. C.;Krogh-Jespersen, K.; Goldman, A. S. Chem. Commun. 1997, 2273-2274. [ Links ]
25. Li, S.; Hall, M. B. Organometallics 1999, 18, 5682. [ Links ] b) Niu, S.; Hall, M. B. J. Am. Chem. Soc. 1999, 121, 3992. [ Links ] c) Krogh-Jespersen, K.; Czerw, M.; Kanzelberger, M.; Goldman, A. S. J. Chem. Inf. Comput. Sci. 2001, 41, 56. [ Links ] d) Krogh-Jespersen, D.; Czerw, M.; Summa, N.; Renkema, K. B.; Achord, P. A.; Goldman, A. S. J. Am. Chem. Soc. 2002, 124, 11404-11416. e) Krogh-Jespersen, K.; Czerw, M.; Goldman, A. S. J. Mol. Catal. A.: Chem. 2002, 189, 95. [ Links ]
26. Jensen, C. M. Chem. Commun. 1999, 2443-2449. [ Links ]
27. Haenel, M. W.; Oevers, S.; Angermund, K.; Kaska, W. C.; Fan, H.-J.; Hall, M. B. Angew. Chem., Int. Ed. 2001, 40, 3596. [ Links ]
28. Dani, P.; Karlen, T.; Gossage, R. A.; Gladiali, S.; van Koten, G. Angew. Chem. Int. Ed. 2000, 39, 743-745. [ Links ]
29. Naota, T.; Takaya, H.; Murahashi, S.-I. Chem. Rev. 1998, 98, 2599. [ Links ]
30. Dani, P.; Albrecht, M.; van Klink, G. P. M.; van Koten, G. Organometallics 2000, 19, 4468. [ Links ]
31. Albrecht, M.; Kocks, B. M.; Spek, A. L.; van Koten, G. J. Organomet. Chem. 2001, 624, 271. [ Links ]
32. Ito, Y.; Sawamura, M.; Hayashi, T. J. Am. Chem. Soc. 1986, 108, 6405-6406. [ Links ]
33. Gorla, F.; Togni, A.; Venanzi, L. M.; Albinati, A.; Lianza, F. Organometallics 1994, 13, 1607. [ Links ] b) Gorla, F.; Venanzi, L. M.; Albinati, A. Organometallics 1994, 13, 43. [ Links ]
34. Zhang, X.; Longmire, J. M.; Shang, M. Organometallics 1998, 17, 4374-4379. [ Links ]
35. Stark, M. A.; Richards, C. J. Organometallics 2000, 19, 1282-1291. [ Links ]
36 Gimenez, R.; Swager, T. J. Mol. Catal. A: Chem. 2001, 166, 265-273. [ Links ]
37 Morales-Morales, D.; Cramer, R. E.; Jensen, C. J. Organomet. Chem. 2002, 654, 44. [ Links ]
38. Longmire, J. M.; Zhang, X. Tetrahedron Lett. 1997, 38, 1725-1728. [ Links ]
39 Morales-Morales, D.; Grause, C.; Kasaoka, K.; Redón, R.Cramer, R. E.; Jensen, C. M. Inorg. Chim. Acta 2000, 300-302, 958-963. [ Links ]
40 Grube, G. H.; Elliott, E. L.; Steffens, R. J.; Jones, C. S.; Baldridge, K. K.; Siegel, J. S. Org, Lett. 2003, 5, 713-716. [ Links ]
41 Wallner, O. A.; Szabó, K. J. Org. Lett. 2004, 6, 1829-1831. [ Links ]
42 Gu, X-Q.; Chen, W.; Morales-Morales, D.; Jensen, C. J. Mol. Catal A. 2002, 189, 119-124. [ Links ]
43 Morales-Morales, D.; Redón, R.; Wang, Z.; Yung, C.; Magnuson, K.; Jensen, C. M. Can. J. Chem. 2001, 79, 823-829. [ Links ]
44. Zhang, X. W.; Fried, A.; Knapp, S.; Goldman, A. S. Chem. Commun. 2003, 2060-2061. [ Links ]
45. Gottker-Schnetmann, I.; White, P.; Brookhart, M. J. Am. Chem. Soc. 2004, 126, 1804-1811. [ Links ]
46 Morales-Morales, D; Redón, R; Yung, C; Jensen, C. M. Inor. Chim. Acta. 2004, 357, 2953-2956. [ Links ]
47. Naghipour, A.; Sabounchei, S. J.; Morales-Morales, D.; Hernández-Ortega, S.; Jensen, C. M. J. Organomet. Chem. 2004, 689, 2494-2502. [ Links ]
48. Meijer, M. D.; Mulder, B.; Van Klink, P. M. G.; van Koten, G. Inorg. Chim. Acta. 2003, 352, 247-252. [ Links ]
49. Koridze, A. A.; Kuklin, S. A.; Sheloumov, A. M.; Kondrashov, M. V.; Dolgushin, F. M.; Peregudov, A. S.; Petrovskiia, P. V. Russ. Chem. Bull., Int. Ed., 2003, 52, 2754-2756. [ Links ]
50. Takenaka, K.; Uozumi, Y. Org. Lett. 2004, ASAP. [ Links ]
51. Melaiye, A.; Simons, R, S.; Milsted, A.; Pingitore, F.; Wesdemiotis, C.; Tessier, C. A.; Youngs, W. J. J. Med. Chem. 2004, 47, 973-977. [ Links ]
52. Guillena, G.; Rodríguez, G.; van Koten, G. Tetrahedron Lett. 2002, 43, 3895-3898. [ Links ]
53. Kleij, A. W.; Gossage, R. A.; Klein Gebbink, R. J. M.; Brinkmann, N.; Reijerse, E. J.; Kragl, U.; Lutz, M.; Spek, A. L.; van Koten, G. J. Am. Chem. Soc. 2000, 122, 12112. [ Links ] b) Knapen, J. W. J.; van der Made, A. W.; de Wilde, J. C.; van Leeuwen, P.W. N.M.; Wijkens, P.; Grove, D. M.; van Koten, G. Nature 1994, 372, 659. [ Links ] c) Kleij, A. W.; Gossage, R. A.; Jastrzebski, J. T. B. H.; Lutz, M.; Spek, A. L.; van Koten, G. Angew. Chem. Int. Ed. 2000, 39, 176. [ Links ]

