










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.3 Ciudad de México jul./sep. 2004
Investigación
Adsorción de Cd(II) en solución acuosa sobre diferentes tipos de fibras de carbón activado
Roberto Leyva Ramos,1* Paola Elizabeth Díaz Flores,1 Rosa María Guerrero Coronado,1 Jovita Mendoza Barrón1 y Antonio Aragón Piña2
1 Centro de Investigación y Estudios de Posgrado, Facultad de Ciencias Químicas, Universidad Autónoma de San Luis Potosí, Av. Dr. Manuel Nava # 6, San Luis Potosí, SLP 78210, México, Tel.: 444-826-2440, Fax: 444-826-2372. E-mail: rlr@uaslp.mx
2 Instituto de Metalurgia, Universidad Autónoma de San Luis Potosí, Av. Sierra Leona No. 550, Lomas 2ª Sección.
Recibido el 15 de abril del 2004.
Aceptado el 5 de agosto del 2004.
Resumen
Se estudió la adsorción de Cd(II) sobre Fibras de Carbón Activado (FCA) en las formas de Tela y Fieltro, sin oxidar y oxidadas con HNO3. Se encontró que el Cd(II) se adsorbió en los sitios ácidos y la FCA-Tela presentó mayor capacidad de adsorción porque contiene mayor concentración de sitios ácidos. La capacidad de adsorción de las FCA aumentó con la oxidación ya que se incrementó la concentración de sitios ácidos. La capacidad de adsorción de las FCA depende de sus propiedades de textura y fisicoquímicas.
Palabras clave: Adsorción, cadmio, fibras de carbón activado.
Abstract
It was studied the adsorption of Cd(II) onto Activated Carbon Fibers (ACF) in the forms of Cloth and Felt, without oxidizing and oxidized with HNO3. It was found that Cd(II) was adsorbed on the acidic sites and that the Cloth had higher adsorption capacity because it contained greater concentration of acidic sites. The adsorption capacity of the ACF increased with the oxidation since the concentration of acidic sites was increased. The adsorption capacity of the ACF depends upon its textural and physicochemical properties.
Key words: Activated carbon fiber, adsorption, cadmium.
Introducción
En los últimos 30 años se ha investigado ampliamente la adsorción de Cd(II) sobre carbón activado y otros adsorbentes como un método para remover Cd(II) presente en soluciones acuosas. La mayoría de los estudios se han enfocado a medir las isotermas de adsorción de Cd(II) sobre carbón activado granular (CAG) y en polvo (CAP), y a evaluar el efecto del pH y de la temperatura en la isoterma de adsorción. En estos estudios se ha reportado que la cantidad de Cd(II) adsorbido aumenta drásticamente incrementando el pH de 2 a 6 [1, 2], que el Cd(II) no se adsorbe sobre carbón activado a pH menores entre 2 y 3 [3] y que la cantidad de Cd(II) adsorbido se reduce disminuyendo la temperatura [4].
Recientemente se ha comercializado una nueva forma de carbón activado que se prepara a partir de la carbonización y activación de telas de fibras de diversos materiales poliméricos, tales como nylon, rayón, celulosa, resina fenólica, poliacrilonitrilo (PAN) y brea de alquitrán [5]. Esta novedosa forma se le conoce como Fibra de Carbón Activado (FCA) y se fabrica en dos presentaciones como tela y fieltro. Las características de las FCA dependen de la tela del material polimérico que se utilizó para producirla.
FCA ofrece varias ventajas en comparación con CAG y CAP. La estructura porosa de la FCA está principalmente constituida por microporos mientras que el CAG y CAP tienen estructura porosa muy compleja formada por microporos, mesoporos y macroporos. Los diámetros de las fibras del FCA (0.006 a 0.017 mm) son en promedio 100 veces menores que los diámetros de las partículas de CAG (1 a 3 mm) y ligeramente menores que los diámetros de las partículas de PAC (0.015 a 0.025 mm). Por lo tanto, la velocidad de adsorción en la FCA es mucho más rápida que en el CAG y ligeramente mas rápida que en el CAP.
Otra ventaja importante de la FCA es que la distancia entre fibra y fibra se puede controlar seleccionando el material precursor, esto permite que la FCA se pueda empacar en adsorbedores de lecho fijo. Es bien sabido que los adsorbedores de lecho fijo no se empacan con CAP porque la caída de presión sería muy alta debido a que el diámetro de la partícula del CAP es muy pequeño. En los procesos en que se utiliza CAP, la velocidad de adsorción es rápida pero presenta la desventaja de que el CAP es muy difícil de manipular y es necesario filtrar la solución para separarlo, esto ocasiona pérdidas y uso ineficientemente del CAP [6]. En el caso de la FCA, la velocidad de adsorción es tan rápida como en el CAP pero no presenta los problemas de manipulación.
La adsorción de cadmio sobre CAG y CAP se ha investigado muy extensamente [1, 3, 7-9] pero muy pocos estudios se han realizado acerca de la adsorción de Cd(II) sobre FCA. Rangel Méndez y Streat [10] estudiaron la adsorción de Cd(II) sobre tela de FCA fabricada a partir de PAN y encontraron que la capacidad de la FCA oxidada por ozonización y electroquímicamente se incrementa hasta 13 veces con respecto a la FCA sin oxidar; sin embargo, la FCA oxidada con ozono presentó daño físico. En un estudio acerca de la adsorción de Cd, Zn y Hg en solución acuosa sobre tela de FCA producida a partir de Rayón, se reportó que la cantidad adsorbida de Cd y Zn se incrementa drásticamente aumentando el pH de la solución mientras que la de Hg varió muy levemente [11]. Hasta la fecha, no se han comparado las capacidades de adsorción de los diferentes tipos y formas de FCA, ni se ha investigado detalladamente la dependencia de la capacidad de adsorción de la FCA con respecto a sus características fisicoquímicas.
El objetivo principal de este trabajo es determinar la isoterma de adsorción de Cd(II) sobre diferentes tipos y formas de fibras y estudiar como afectan las características superficiales de las fibras a la capacidad de adsorción de la FCA.
Metodología experimental
Fibras de carbón activado
En este estudio se usaron dos formas de FCA que se conocen comercialmente como: AM1132 (Fieltro) y AW1102 (Tela). En el presente trabajo estas dos formas se referirán como FCA-Tela y FCA-Fieltro. Estas FCA son fabricadas por KoThmex a partir de poliacrilonitrilo (PAN). Las FCA tal como se recibieron por parte del fabricante se lavaron repetidas veces con agua destilada para remover polvo adherido y después se secaron en una estufa a 110°C durante 24 h.
Antes de realizar los experimentos de adsorción, las FCA se pretrataron para eliminar el aire que pudiera quedar atrapado dentro de los poros cuando se sumergieron en solución acuosa. El objetivo del pretratamiento es lograr que casi todos los poros de la FCA se llenen con agua desionizada para asegurar que la mayor parte del área interna de la FCA este disponible para la adsorción. El pretratamiento se describe a continuación: Se pesó una masa definida de la FCA y luego se sumergió en agua desionizada hirviendo durante 15 min. La FCA se dejó enfriar dentro del agua y se removió hasta que se colocó dentro de la solución del adsorbedor.
Oxidación de las FCA
Las FCA se oxidaron por un procedimiento similar al reportado por Bandosz y cols. [12]. En un vaso de precipitados de 1 L se agregaron 20 g de FCA y 500 mL de una solución 10% (v/v) de HNO3, preparada con HNO3 concentrado y agua desionizada. La solución ácida conteniendo la FCA se calentó durante 2 h en una parrilla eléctrica, procurando que la temperatura no excediera 50°C. La solución ácida que contenía la FCA se dejó enfriar y luego se drenó. Enseguida, la FCA oxidada se lavó varias veces con agua desionizada hasta que el pH del agua de lavado ya no varió. La FCA se secó en una estufa a 110°C durante 24 h y se guardó en un recipiente seco y cerrado.
Determinación de sitios activos
Los sitios activos de las FCA se determinaron por el método de titulación ácido-base propuesto por Boehm [13]. Los sitos ácidos se neutralizaron con una solución patrón 0.1 M de NaOH y los sitios básicos con una solución patrón 0.1M de HCl.
Los sitios activos se determinaron aforando un matraz volumétrico de 50 mL con solución neutralizante 0.1 N y agregando aproximadamente 1 g de FCA. El matraz se sumergió parcialmente en un baño de temperatura constante a 25°C y se dejó por 5 días. El matraz se agitó manualmente 2 veces al día. Una vez transcurrido este tiempo, se tomó una alícuota de 10 mL y se tituló con soluciones 0.1 N de HCl o NaOH, según sea el caso [12].
Determinación del punto de carga cero
El punto de carga cero (PCC) de la FCA se determinó por un procedimiento muy similar al reportado por Babic y col. [11], que se describe a continuación. A un vaso de precipitados se añadieron 0.1 g de FCA molida finamente en un mortero de Ágata y 20 mL de una solución 0.01 M de KCl-0.004 M de KOH. La solución se mantuvo en agitación continua durante 48 h. Enseguida, se realizó la titulación con una solución de HCl 0.1M usando una bureta y en una corriente de nitrógeno para evitar que el dióxido de carbono presente en el aire se absorba en la solución y se formen CO3-2 y HCO3-1. La solución titulante se adicionó lentamente, 0.1 mL por adición y se registró el volumen agregado y el pH de la solución. Por otro lado, se realizó la valoración de la solución de 0.01M de KCl -0.004 de KOH bajo las mismas condiciones pero sin FCA. El PCC de la FCA se determinó graficando el pH de la solución contra el volumen de la solución titulante para la solución sin FCA y para la solución con FCA, el pH en donde estas dos curvas se interceptan corresponde al PCC. Otra forma de interpretar los datos experimentales es calcular la carga de la superficie de la FCA usando las ecuaciones reportadas por Babic y col. [11], en este caso el pH al cual la carga de la superficie es cero corresponde al PCC.
Caracterización de las FCA
El área superficial interna, el diámetro promedio de los poros y el volumen promedio de los poros se determinaron por medio de un equipo de fisisorción, marca Micromeritics, modelo ASAP 2010, que se basa en el método de adsorción de N2 a una temperatura cercana a su punto de ebullición (método BET).
La superficie de las FCA se analizó por medio de un microscopio electrónico de barrido, marca Leica-Cambridge, modelo S420-i, equipado con un sistema de microanálisis Link/ISIS-OXFORD de energía dispersa (EDS). El análisis elemental de las FCA oxidadas y sin oxidar se realizó utilizando un Analizador Elemental, marca CE Instruments, modelo EA 1110 que determina las fracciones en peso de nitrógeno, hidrógeno, oxígeno y carbón presentes en la muestra.
Determinación de la concentración de Cd(II)
La concentración de Cd(II) en solución acuosa se determinó por espectrofotometría de absorción atómica, midiendo la absorbancia de cada muestra en un espectrofotómetro de absorción atómica de doble haz, marca Varian, modelo SpectrAA-20, y calculando su concentración por medio de una curva de calibración que se preparó previamente determinando la absorbancia de 4 ó 5 soluciones patrón. El espectrofotómetro se calibró a cero utilizando agua desionizada como blanco.
Obtención de datos experimentales de las isotermas de adsorción
Los datos experimentales de las isotermas de adsorción se obtuvieron en un adsorbedor de lote que constó de un matraz Erlenmeyer de 500 mL, al cual se agregaron 480 mL de una solución de Cd(II) de concentración conocida. Una cierta masa de FCA se introdujo dentro de la solución del adsorbedor, y este último se sumergió parcialmente en un baño de temperatura constante. La solución se mezcló por medio de una barra de agitación magnética recubierta de Teflón, accionada por una placa de agitación que se encontraba debajo del baño de temperatura constante. La solución y la FCA se dejaron en contacto hasta que se alcanzó el equilibrio. Cada determinado tiempo se tomó una muestra de la solución y se determinó la concentración de Cd(II). Se consideró que se había alcanzado el equilibrio cuando la concentración de dos muestras sucesivas ya no varió significativamente. El equilibrio se alcanzó de 5 a 7 días. La masa de Cd(II) adsorbido se calculó por medio de un balance de masa.
Discusión de resultados
Características fisicoquímicas y de textura de las FCA sin oxidar
El área superficial, volumen de poros y el diámetro promedio de los poros se exhiben en la Tabla 1 para las FCA sin oxidar. En esta tabla se nota que las áreas superficiales de la FCA-Tela y de la FCA-Fieltro sin oxidar son 1090 y 1220 m2/g y los volúmenes de los poros son de 0.53 y 0.60 cm3/g, respectivamente. FCA fabricadas a partir de celulosa, brea de alquitrán y poliacrilonitrilo (PAN) presentaron áreas superficiales y volúmenes de poro similares a los valores reportados en este trabajo [5, 14].
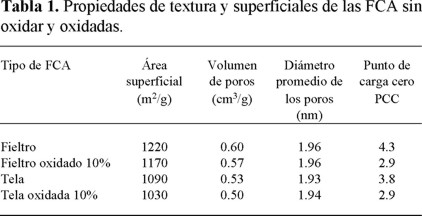
El diámetro promedio de los poros de las FCA es inferior a 2 nm, esto revela que gran parte de la estructura porosa de las FCA esta constituida principalmente por microporos, esto coincide con lo reportado por otros autores [14-16].
En la Figura 1 se muestran las concentraciones de los sitios activos de las FCA sin oxidar y se observa que la concentración de sitios ácidos es mayor que la de básicos en ambos tipos de FCA. Las concentraciones de sitios ácidos en la FCA-Tela y FCA-Fieltro son 1.055 y 0.617 meq/g y las de sitios básicos son 0.296 y 0.444 meq/g, respectivamente. Esto indica claramente que la superficie de las FCA es de carácter ácido.

En la Figura 2 se muestra la carga de la superficie de la FCA-Fieltro y se señala el punto de carga cero (PCC). Las concentraciones de H+ y OH- adsorbidos sobre la superficie son iguales en el PCC y por lo tanto, la carga de la superficie es neutra [17]. La distribución de la carga de la superficie de la FCA-Tela es muy similar a la FCA-Fieltro y por ésta razón no se presenta en forma gráfica. Los valores del PCC para las FCA se encuentran en la Tabla 1.

En la Figura 2 se observa que la carga superficial del carbón es positiva para valores de pH menores al PCC, neutra cuando el pH es igual al PCC y negativa para valores de pH mayores al PCC. Este comportamiento es típico de los carbones activados y de las FCA, y ha sido reportado en varios trabajos [18-20]. Los valores de PCC son 3.8 y 4.3 para la FCA-Tela y la FCA-Fieltro, respectivamente, esto se debe a que la concentración de sitios ácidos en ambas FCA es mayor que la de los sitios básicos. Estos valores corroboran que ambos tipos de FCA son de carácter ácido y se encuentran por debajo de los reportados por Rangel Mendez y Streat [10] para una FCA fabricada también de PAN. Por otro lado, el PCC de la FCA-Tela es mas ácido que el PCC de la FCA-Fieltro ya que la FCA-Tela tiene mayor proporción de sitios ácidos con respecto a los sitios básicos.
Oxidación de las FCA con solución de HNO3
En este trabajo la superficie de las FCA se modificó por medio de oxidación con una solución 10% de ácido nítrico para investigar como afecta la oxidación a sus propiedades fisicoquímicas y a su capacidad de adsorción.
Los resultados de la Tabla 1 revelan que la oxidación causó una ligera disminución en el área superficial y en el volumen de poro, mientras que el diámetro promedio de los poros aumentó muy ligeramente. Este comportamiento se puede atribuir a la destrucción o combustión de parte de las paredes de los poros, y al bloqueo de poros estrechos ocasionado por los productos formados durante la oxidación o por la introducción de grupos oxigenados en la superficie [10, 19].
En la Figura 1 se puede observar de manera gráfica como se incrementaron las concentraciones de los sitios ácidos y se redujeron las de los sitios básicos en ambas FCA cuando se oxidaron con HNO3. Las concentraciones de los sitios ácidos aumentaron casi al doble, la de los sitios básicos disminuyeron a cerca de la mitad y la concentración total de los sitios activos aumentaron alrededor de 62%. Varios investigadores [10, 19, 21] han encontrado que la oxidación química de CAG y FCA con solución de HNO3 incrementa la concentración de sitios ácidos, mientras que disminuye la de los sitios básicos. Este comportamiento se debe a la introducción de grupos oxigenados que le confieren a la superficie un carácter ácido.
En la Tabla 1 se nota que el PCC de las FCA disminuyó con la oxidación, esto se atribuyó a que en la oxidación se introdujeron grupos oxigenados en la superficie incrementando la concentración de sitios ácidos. Para la FCA-Fieltro la diferencia entre el PCC de la FCA sin oxidar y el de la oxidada es mayor que para la FCA-Tela. El PCC de la FCA-Fieltro y de la FCA-Tela disminuyeron 32% y 23%, respectivamente, esto se debió a que en la oxidación se incrementaron mucho más los sitios ácidos en la FCA-Fieltro que en la FCA-Tela.
Análisis por microscopía electrónica de barrido
Las FCA-Fieltro sin oxidar se muestran en la Figura 3 y se observa claramente que no existe orden en el arreglo de las fibras. Además, se nota que cada fibra está formada por varios filamentos del material precursor original y el número de filamentos es variable, y el diámetro promedio de los filamentos es de 3 µm.

Las fotomicrografías de la FCA-Tela sin oxidar se exhiben en la Figura 4. En este caso, el arreglo de las fibras es como de un material textil porque el material precursor era tela de fibras de PAN. A diferencia de las FCA-Fieltro, las fibras de la FCA-Tela están formadas solamente por un filamento y el diámetro de la fibra es de 5 µm. No se muestran fotomicrografías de FCA oxidadas porque se notó que la oxidación con solución 10% de HNO3 no modificó la morfología de la FCA.

Composición química elemental de las FCA
En la Tabla 2 se muestran los resultados del análisis elemental de las FCA sin oxidar y los resultados indican que las FCA están principalmente constituidas de Carbón y Oxígeno y contienen en mucha menor proporción Nitrógeno e Hidrógeno. Esto era de esperarse ya que ambos tipos de FCA se prepararon a partir de tela fabricada de PAN. La oxidación de ambos tipos de FCA ocasionó que las cantidades de oxígeno e hidrógeno se incrementaron mientras que la cantidad de carbón disminuyó. Lo primero se debe a que al oxidar la FCA se introducen grupos ácidos que contienen oxigeno e hidrógeno. El segundo efecto se explica recordando que en la oxidación se consume parte de la estructura de la fibra, esto se corroboró con la disminución del área superficial y volumen de poros causadas por la oxidación (Ver Tabla 1).

Isoterma de adsorción
Los datos experimentales de las isotermas de adsorción de Cd(II) sobre las FCA se ajustaron usando los modelos de las isotermas de Langmuir y Freundlich que se representan matemáticamente como:

Los valores de las constantes de estas isotermas se estimaron usando un método de mínimos cuadrados. Los valores de las constantes, así como los porcentajes de desviación promedio se exhiben en la Tabla 3. Los porcentajes de desviación variaron de 5.10 a 27.6% para la isoterma de Langmuir y de 7.50 a 10.7% para la isoterma de Freundlich. Se consideró que la isoterma de Freundlich ajustó mejor los datos que la de Langmuir ya que la primera presentó menor porcentaje de desviación en tres de los cuatro casos (Véase Tabla 3).
Efecto del tipo de FCA sobre la adsorción de Cd(II)
En la Figura 5 se graficaron las isotermas de adsorción de Cd(II) en solución acuosa sobre las FCA sin oxidar, sin ajuste de pH y a T=25°C, y se observa que la FCA-Tela posee mayor capacidad de adsorción que la FCA-Fieltro. Está bien documentado en la literatura que los cationes metálicos de Cd(II) en solución acuosa se adsorben sobre los sitios ácidos del carbón activado [22] y que los principales sitios ácidos son los carboxílicos, fenólicos y lactónicos. Esto explica el resultado anterior ya que la FCA-Tela tiene mayor concentración de sitios ácidos en comparación con la FCA-Fieltro.

Es importante mencionar que el pH de la solución juega un papel importante en la adsorción de Cd(II); sin embargo, el pH de la solución no se mantuvo constante ya que disminuyó muy levemente durante la adsorción de Cd(II) sobre FCA. En promedio el pH de la solución de Cd(II) se redujo de 4.3 a 4.0 que se debió a la transferencia de iones H+ de la FCA a la solución. Se considera que esta disminución modifica muy ligeramente los resultados que se presentan en la Figura 5.
Las capacidades de adsorción de las FCA sin oxidar se pueden comparar calculando la masa de Cd(II) adsorbido a una determinada concentración de Cd(II) en el equilibrio. Las masas de Cd(II) adsorbido a una concentración en el equilibrio de 50 mg/L, Q50, son 4.16 y 3.56 mg/g, para la FCA-Tela y FCA-Fieltro, respectivamente. La capacidad de adsorción de la FCA-Tela es en promedio 17% mayor que la FCA-Fieltro; sin embargo, la concentración de los sitios ácidos en la FCA-Tela es 70% mayor que en la FCA-Fieltro. Esto indica que el Cd(II) se adsorbe selectivamente sobre algún tipo de sitios ácidos, de otra forma la capacidad de adsorción de la FCA-Tela sería 70% mas grande que la de la FCA-Fieltro.
Efecto de la oxidación en la capacidad de adsorción de las FCA
Las isotermas de adsorción de Cd(II) sobre ambas FCA oxidadas se exhiben en la Figura 6 y se observa que la capacidad de las FCA para adsorber Cd(II) aumenta al oxidarlas con solución 10% de HNO3. El aumento de la capacidad de adsorción se puede explicar recordando que la concentración de sitios ácidos en las FCA se incrementó en la oxidación y que el Cd(II) se adsorbe principalmente sobre los sitios ácidos.

Las capacidades de adsorción de las FCA oxidadas se pueden comparar calculando las masas de Cd(II) adsorbido a una misma concentración en el equilibrio. Para una concentración en el equilibrio de 50 mg/L, la masa de Cd(II) adsorbido, Q50, es 11.9 mg/g sobre la FCA-Tela oxidada 10% y de 4.16 mg/g sobre la FCA-Tela sin oxidar. Esto corresponde a un aumento de la capacidad de adsorción de cerca de 3 veces. Además, la capacidad de adsorción de la FCA-Tela oxidada es en promedio, 52% mayor que la FCA-Fieltro oxidado. Esto se debe nuevamente a que la FCA-Tela oxidada tiene mayor concentración de sitios ácidos que la FCA-Fieltro oxidado.
Conclusiones
La caracterización por fisisorción de nitrógeno reveló que las FCA tienen áreas superficiales entre 1000 y 1200 m2/g. Los poros de las FCA estudiadas son predominantemente microporos. El área superficial de las FCA se reduce ligeramente durante la oxidación debido a la destrucción de las paredes de los poros y al bloqueo de poros estrechos ocasionado por los productos de la oxidación o por la introducción de grupos oxigenados.
Los puntos de carga cero (PCC) de la FCA-Tela y la FCA-Fieltro son 3.8 y 4.3, respectivamente. Estos PCC son ácidos y se debe a que las FCA poseen una mayor concentración de sitios activos ácidos que de sitios básicos. Los PCC de la FCA-Tela y FCA-Fieltro oxidados al 10% son 2.9 y 2.9, respectivamente. El PCC de las FCA disminuyó con la oxidación porque se incrementó la concentración de sitios ácidos.
El análisis elemental mostró que las FCA están principalmente constituidas por Carbón y Oxígeno, y contienen en mucha menor proporción Nitrógeno e Hidrógeno. En ambas formas de FCA las cantidades de Oxígeno e Hidrógeno se incrementaron durante la oxidación mientras que la cantidad de carbón disminuyó. Esto se debió a la introducción de grupos ácidos que contienen Oxígeno e Hidrógeno.
La FCA-Fieltro no exhibió orden en el arreglo de las fibras y cada fibra está formada por varios filamentos que tienen diámetro promedio de 3 µm. La FCA-Tela presentó un arreglo tipo material textil y cada fibra está constituida por un solo filamento de diámetro de 5 µm. La oxidación de las FCA con solución 10% de HNO3 no modificó su morfología.
La FCA-Tela presentó mayor capacidad para adsorber Cd(II) que la FCA-Fieltro, esto se debió a que la primera tenía mayor concentración de sitios ácidos y que el Cd(II) se adsorbió principalmente en este tipo de sitios. La capacidad de la FCA-Tela para adsorber Cd(II) aumentó 2.9 veces cuando se oxidó con HNO3. La capacidad de la FCA-Tela oxidada fue 52% mayor que la de la FCA-Fieltro oxidado.
Referencias
1. Reed, B. E.; Matsumoto, M. R. Sep. Sci. Technol. 1993, 28, 2179-2195. [ Links ]
2. Leyva Ramos, R.; Rangel Méndez, J. R.; Mendoza Barrón, J.; Fuentes Rubio, L.; Guerrero Coronado, R. M. Water Sci. Technol. 1997, 35, 205-211. [ Links ]
3. Bhattacharya, A. K.; Venkobachar, C. J. Environ. Eng. 1984, 110, 110-122. [ Links ]
4. Marzal, P.; Seco, A.; Gabaldón, C. J. Chem. Tech. Biotechnol. 1996, 66, 279-285. [ Links ]
5. Suzuki, M. Carbon 1994, 32, 577-586. [ Links ]
6. Cheremisinoff, P. N.; Cheremisinoff, N. P. Carbon Adsorption for Pollution Control. Editorial Prentice Hall, USA, 1993, 21. [ Links ]
7. Huang, C. P.; Wirth, P. K. J. Environ. Eng. Div. 1982, 108 (EE6), 1280-1299. [ Links ]
8. Reed, B. E.; Nonavinakere, S. K. Sep. Sci. Tech. 1992, 27, 1985-2000. [ Links ]
9. Reed, B. E.; Arunachalam, S. Environ. Prog. 1994, 13, 60-64. [ Links ]
10. Rangel Méndez, J. R.; Streat, M. Water Res. 2002, 36, 1244-1252. [ Links ]
11. Babic, B. M.; Milojic, S. M.; Polovina, M. J.; Kaludjerovic, B. V. Carbon 1999, 37, 477-481. [ Links ]
12. Bandosz T. J.; Jagiello J.; Schwarz, J. Anal. Chem. 1992, 64, 891-895 [ Links ]
13. Boehm, H. P. Chemical Identification of Surface Groups. Heidelberg, Germany. 1970. [ Links ]
14. Li, Z.; Kruk, M.; Jaroniec, M.; Ryu, S-K. J. Colloid and Interface Sci. 1998, 204, 151-156. [ Links ]
15. Le Cloirec, P.; Brasquet, C.; Subrenat, E. Energy & Fuels. 1997, 11, 331-336. [ Links ]
16. Carrott, J. A.; Nabais, J. M. V.; Ribeiro Carrott, M. M. L. Carbon 2001, 39, 1543-1555. [ Links ]
17. Bockris, J. O. M.; Otagawa, T. J. Electrochem. Soc.1984, 131, 290-302. [ Links ]
18. Minguang, D. J. Colloid and Interface Sci. 1994, 164, 223-228. [ Links ]
19. Shim, J-W.; Park, S-J.; Ryu, S-K. Carbon 2001, 39, 1635-1642. [ Links ]
20. Babic, B. M.; Milonjic, S. M.; Polovina M. J.; Cupic, S.; Kaludjerovic, B. V. Carbon 2002, 40, 119-115. [ Links ]
21. Leyva-Ramos, R.; Bernal-Jácome, L. A.; Mendoza-Barrón, J.; Fuentes-Rubio, L.; Guerrero-Coronado, R. M. J. Hazard. Mater. 2002, B90, 27-38. [ Links ]
22. Radovic, L. R.; Moreno Castilla, C.; Rivera Utrilla, J. In: Chemistry and Physics of Carbon, Radovic, L. R., Ed., Marcel Dekker, New York, 2001, 256-262. [ Links ]

