










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.1 Ciudad de México ene./mar. 2004
Investigación
Síntesis de sesquiterpenos isocariolánicos con potencial actividad antifúngica frente a Botrytis cinerea
Juan Carlos Racero,1* Isidro González Collado2 y Antonio José Macías2
1 Laboratorio de Síntesis Orgánica. Facultad de Ciencias Químicas. Universidad Autónoma de San Luis Potosí. Avda. Dr. Manuel Nava No.6. 78210 San Luis Potosí, SLP. México. Tel: (444) 826 24 40. Fax: (444) 826 23 72. E-mail: jcracero@uaslp.mx
2 Departamento de Química Orgánica. Facultad de Ciencias. Universidad de Cádiz. República Saharaui s/n. 11510 Puerto Real, Cádiz. España.
Recibido el 14 de enero del 2004.
Aceptado el 13 de abril del 2004.
Resumen
Los sesquiterpenos con esqueleto de isocariolano representan potenciales compuestos antifúngicos debido a que poseen analogía estructural con los metabolitos fitotóxicos de Botrytis cinerea. En este estudio se presenta la preparación de isocariolanos, funcionalizados en el C-8 y C-9, por reagrupamiento de derivados del cariofileno y los reagrupamientos pinacolínicos de esos derivados isocariolánicos bajo condiciones de Mitsunobu. Finalmente se presentan los resultados in vitro de actividad antifúngica frente a Botrytis cinerea. El compuesto 8 presentó una interesante actividad frente al citado hongo.
Palabras clave: Sesquiterpeno, isocariolano, reagrupamiento, Mitsunobu, antifúngico, Botrytis cinerea.
Abstract
Sesquiterpenes with isocaryolane skeleton represent potential antifungal compounds because they possess a structural similarity to the phytotoxic metabolites produced by Botrytis cinerea. In this paper we report the preparation of isocaryolane, with functionality at C-8 and C-9, by rearrangement of caryophylene derivatives and pinacol rearrangements of these isocaryolane derivatives under Mitsunobu conditions. Finally we report in vitro results of antifungal activity against Botrytis cinerea. Compound 8 showed an interesting activity against this fungus.
Keywords: Sesquiterpene, isocaryolane, rearrangement, Mitsunobu, antifungal, Botrytis cinerea.
Introducción
Escasos son los antecedentes acerca de la obtención de compuestos con esqueleto de isocariolano. La primera vez que se reportó un compuesto con este esqueleto fue en 1955 [1]. El esqueleto se obtuvo partiendo de un aducto de cariofileno con anhídrido maleico mediante la reacción de ambos en benceno a reflujo. Este aducto se reagrupaba fácilmente con reactivos electrofílicos dando lugar a compuestos con esqueleto tricíclico cuyas estructuras entonces no pudieron ser confirmadas con exactitud. La obtención de la amida derivada a partir del ácido carboxílico obtenido en el reagrupamiento anterior permitió determinar la estructura exacta mediante un análisis por difracción de rayos X. Estudios posteriores sobre ciclación de distintos derivados con esqueleto de cariofileno mediante procesos de oximercurización-desmercurización [2] permitieron obtener nuevamente derivados con esqueleto de isocariolano. Partiendo del 4β,5α- y 4β,5β epoxicariofileno se obtuvieron los correspondientes hidroxiésteres que, por desacetilación, condujeron a los isocariolano-8,9-dioles. Posteriormente, en estudios acerca de la ciclación de óxido de cariofileno en medio ácido y empleando Al2O3 como catalizador se obtuvo, como uno de los productos de la reacción, el isocariolan-8-ol [3]. El producto de eliminación o deshidratación, el isocario-lan-8(9)-eno [4], se obtuvo partiendo de un derivado dihidroclorado del cariofileno.
El esqueleto de isocariolano presenta características estructurales sumamente interesantes y atractivas. Este hecho, unido a los escasos datos que existen acerca de su reactividad, así como su analogía con los esqueletos de clovanos y cariolanos (activos frente a Botrytis cinerea) [5], nos llevó a abordar el estudio del comportamiento químico de estos compuestos.
Desde los años 90 nuestro grupo de investigación viene trabajando en el diseño biosintético de fungicidas, mediante estudios de ciclaciones y reagrupamientos partiendo del 4β,5α-epoxicariofileno 1 en presencia de tetracianoetileno (TCNE). El TCNE es un catalizador àcido π que forma un complejo de transferencia de carga, a partir del cual se producen diversos reagrupamientos en su esqueleto. El proceso de formación del complejo de transferencia de carga entre el TCNE [6] y una molécula con facilidad de donar electrones (óxido de cariofileno) transcurre a través de la transferencia de un electrón desde el orbital no enlazante del oxígeno del epóxido, hacia el sistema π del catalizador. Dicho complejo de transferencia provoca un defecto de carga sobre el oxígeno dando lugar a una deficiencia de carga en la posición C4. Un posible ataque del doble enlace exocíclico, tanto por la cara inferior como por la superior de la molécula, sobre la posición C4 , con apertura concertada del anillo oxiránico, conduciría al producto de ciclación transanular correspondiente. La formación de los distintos esqueletos está íntimamente relacionada con la conformación más estable adoptada por el producto de partida. En los estudios previos acerca de esta reacción llevados a cabo por nuestro grupo de investigación [7], se obtuvieron tres productos mayoritarios: el alcohol alílico 2, el producto de la alcohólisis 3 y el éter de clovan-2β,9α-diol 4.
Resultados y Discusión
En el estudio mencionado sobre la reacción de óxido de cariofileno catalizada por TCNE [7], se detectó, además de los productos mayoritarios, la presencia de productos minoritarios cuyo esqueleto podría ser de interés, ya que podrían facilitar información sobre el curso de la reacción. Con el objeto de aislar y caracterizar dichos productos se optimizó esta reacción buscando mejorar rendimientos. Tras ensayar diversas condiciones de reacción [6] se obtuvieron nuevamente, 2β-metoxiclovan-9α-ol 4, el producto de alcohólisis 3 y el de eliminación 2 como productos mayoritarios, junto con los productos minoritarios cuyas estructuras resultaron ser: 8β-metoxi-5α-hidroxicariofil-3(4)-eno 5 e isocariolan-9-ona 6 (Fig. 1). La presencia del compuesto 6 en la mezcla de reacción pone de manifiesto la existencia de un tipo de reagrupamiento desconocido hasta ahora en el óxido de cariofileno 1. De acuerdo con los antecedentes bibliográficos [8], la isocariolan-9-ona 6 se obtendría a partir del cariofila-4(12)-8(13)-dien-5α-ol 7, previa formación de un complejo de transferencia sobre el metileno exocíclico C8-C13 y posterior ciclación transanular seguida de transferencia de hidruro (Fig. 2).

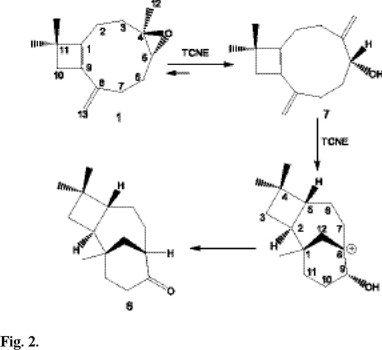
Es importante destacar que, bajo las condiciones de reacción, se produce un novedoso e interesante desplazamiento 1,2 de hidruro que conduce finalmente al producto 6.
El mecanismo de reacción propuesto fue confirmado cuando el cariofila-4(12)-8(13)-dien-5α-ol 7 fue tratado con TCNE en metanol a temperatura ambiente, obteniéndose la isocariolan-9-ona 6, el 8β-metoxi-isocariolan-9α-ol 8 y el aldehido 9 (Figs. 3 y 4).


La estructura de los productos fue establecida por métodos espectroscópicos. El compuesto 8 presenta una banda en IR a 3381 cm-1 característica de un grupo hidroxilo, señales en el espectro de 1H NMR a δ 3.5 ppm (dd, 1H, J = 5.8,11.2 Hz, H-9) correspondiente a un hidrógeno geminal a un grupo hidroxilo y otra señal a 3.21 ppm (s, 3H, H-16) correspondiente a los tres hidrógenos de un grupo metoxilo. Estos datos junto con los experimentos bidimensionales COSY y HETCOR nos permitió proponer una estructura de 8-metoxi-isocariolan-9-ol. Finalmente la orientación del grupo metoxilo pudo ser determinada mediante el estudio del efecto n.O.e positivo observado al irradiar la señal correspondiente al H-8. Los valores de las constantes de acoplamiento del hidrógeno geminal al grupo hidroxilo son característicos de un protón ecuatorial, lo que confirma la orientación axial de dicho grupo hidroxilo. La estructura del compuesto 9 se determinó por difracción de rayos X de la 2,4-dinitrofenilhidrazona derivada [9], ante la imposibilidad de asignar completamente las señales entre 1.2 y 1.9 ppm correspondiente a 14 hidrógenos en el espectro de RMN 1H y dar una estructura del compuesto 9. Los datos cristalográficos confirmaron que se trata del (1S,2S,5R,8S)-1,4,4-trimetiltriciclo [6.2.1.02,5]undecano-8-carbaldehido 9 (Fig. 5).

La presencia en la mezcla de reacción de los compuestos 8 y 9 confirmó el mecanismo de reacción que se había propuesto para la formación de la isocariolan-9-ona 6, por el ataque del doble enlace exocíclico C4-C12 sobre el carbono C8, previa formación de un complejo de transferencia de carga entre el TCNE y el doble enlace exocíclico C8-C13. Este ataque con la consiguiente ciclación transanular implicaba la formación de un carbocatión cabeza de puente en la posición C8 del esqueleto de isocariolano (fig. 3) [10].
A partir de los datos obtenidos y atendiendo a los antecedentes bibliográficos, parece evidente que el esqueleto de isocariolano se obtiene a partir de derivados de cariofiladieno. Las especiales características del esqueleto de cariofileno, con un anillo de nueve miembros dotado de una amplia movilidad y diversas conformaciones, así como la presencia de los dos metilenos exocíclicos, inducen a pensar que la reactividad y el curso de la reacción podrían estar afectados por las condiciones de reacción y por los sustituyentes presentes.
Dada las especiales características de los ácidos π (TCNE) podría pensarse en importantes cambios en los productos de reacción, cuando ésta se llevase a cabo en medio ácido prótico y en medio ácido de Lewis. Cuando el cariofila-4(12),8(13)-dien-5α-ol 7 se sometió a tratamiento en medio ácido a baja temperatura, se obtuvieron los compuestos 6, 9 y el isocariolano-8β,9α-diol 10, resultados consecuentes con el mecanismo propuesto en la figura 3. Cuando el 8β-metoxi-isocariolan-9β-ol 8 fue tratado con HBr (47%) en acetona, con el fin de estudiar su comportamiento en este medio, se obtuvieron dos productos de reacción mayoritarios: la cetona 6 fruto de la migración 1,2 del hidruro y el carbaldehído 9. Por otro lado, cuando el isocariolandiol fue tratado en condiciones ácidas se obtuvieron resultados idénticos a los obtenidos con el derivado metoxilado 8, es decir: 1R,2S,5R, 8S-8-carbaldehído-1,4,4-trimetiltriciclo [6.2.1.02,5] undecano 9 e isocariolan-9-ona 6. Este resultado está de acuerdo con el mecanismos propuesto según el cual en medio ácido el intermedio carbocatiónico más estable es aquel en el que la carga positiva queda localizada sobre el C8.
Atendiendo a los productos obtenidos con los derivados clovánicos [8], los compuestos 8 y 10 se sometieron a reacción bajo condiciones de Mitsunobu modificadas [11] con el fin de estudiar su comportamiento químico. El compuesto 8 fue tratado con trifenilfosfina y dietilazodicarboxilato (DEAD) obteniéndose tras separación cromatográfica en columna y HPLC, un producto mayoritario: triciclo [7.2.1.02,5]-1,4,4-trimetildodecan-8-ona 11 (52%) y recuperándose el producto de partida 8 (25%) [12] (Fig. 6).

Dada la complejidad del espectro de RMN-1H y de los experimentos bidimensionales COSY y HETCOR de la cetona 11 se hizo necesaria la obtención de derivados que ayudaran a la elucidación estructural. De este modo, el producto 11 fue sometido a reducción con NaBH4 obteniéndose dos alcoholes epímeros: 12 (51%) y 13 (15%). El compuesto 12, tras su recristalización en metanol, fue sometido a análisis de difracción de rayos X [9], del que se infirió que se trataba del 1,4,4-trimetiltriciclo [7.2.1.02,5]-dodecan-8β-ol 12, confirmando por tanto que la cetona 11, de la cual procede por reducción, presenta una estructura de 1,4,4-trimetiltriciclo [7.2.1.02,5]-dodecan-8-ona 11. Por otro lado, el tratamiento del isocariolan-8β,9α-diol 10 con DEAD y Φ3P dio como productos mayoritarios la isocariolan-9-ona 6 y el 1,4,4-trimetiltriciclo [7.2.1.02,5]-dodecan-8-ona 11 (Fig. 6). La obtención de la cetona 11 a partir del isocariolanol 8 puede ser explicada en término de un novedoso reagrupamiento pinacolínico descrito por nuestro grupo de investigación. De igual modo, los compuestos 6 y 11 pueden ser explicados mediante un reagrupamiento pinacolínico a partir del diol vecinal 10, siendo sorprendente que dicho reagrupamiento se produzca bajo las condiciones de reacción de Mitsunobu. Se ha propuesto por tanto un mecanismo de reacción que implicaría la salida del ión trifenil fosfonio sobre C9, promovido por un reagrupamiento de Wagner-Meerwein del enlace con disposición antiperiplanar C12-C8 hacia el C9, y posterior ataque del anión hidrazina al ión oxonio sobre C8 dando como producto mayoritario la cetona 11. La obtención de la cetona 6 quedaría justificada tras formación del ión trifenilfosfonio en posición C8, el cual evoluciona mediante transferencia de hidruro 1,2 para dar la cetona 6. Con objeto de confirmar el papel del DEAD en el transcurso de la reacción, se realizaron varios ensayos cambiando las condiciones iniciales empleando como base los estudios de Loibner [13]. Así, cuando se añadió CH2Cl220, y se empleó DEAD en cantidades catalíticas obtuvimos como productos mayoritarios de la reacción el 9β-cloro-8β-metoxi-isocariolano 14, producto de la inversión de la configuración en el C9 por el ataque de un anión cloruro y el 9α-cloro-8β-metoxi-isocariolano 15. En ningún caso se detectó la presencia del producto de reagrupamiento 11. Posteriormente, cuando el experimento se realizó utilizando DEAD en exceso (2.2 eq) aparecieron trazas del producto de reagrupamiento 11 (4%), junto con los productos 14 (18%) y 15 (15%) (Fig. 7). El compuesto 15 fue caracterizado por técnicas espectroscópicas y por difracción de rayos X [9] (Fig. 8).


Estos resultados confirman la hipótesis propuesta y el papel que juega el DEAD en las reacciones pinacolínicas anteriores. La presencia de diclorometano consume el anión de la dietoxicarbonilhidrazina presente en el medio de reacción y genera el anión Cl- que provoca la reacción de sustitución nucleofílica dando los productos 14 y 15.
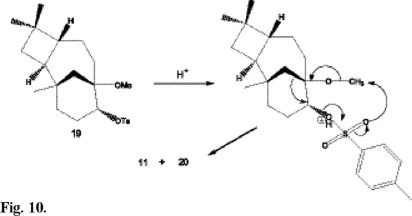
Por último, el papel del DEAD en la reacción descrita, en ausencia o presencia de CH2Cl2, fue confirmado cuando el isocariolan-9α-ol 16 fue sometido a tratamiento bajo condiciones de Mitsunobu obteniéndose como producto de reacción la dietoxicarbonilhidrazina 17 derivada por ataque nucleofílico en la posición C9, mientras que al añadir CH2Cl2 en las condiciones de reacción se obtuvo el 9β-cloroisocariolano 18 (Fig. 9). Los resultados y reacciones descritas hasta ahora indican que el reagrupamiento pinacolínico podría ser orientado mediante la utilización de un buen grupo saliente sobre C8 o sobre C9. Con objeto de confirmar este hecho, el 8β-metoxi-isocariolan-9α-ol 8, el cual conducía a la cetona 6 y el aldehído 9 en condiciones ácidas, fue tratado con cloruro de p-toluensulfonilo obteniéndose como producto mayoritario el 8β-metoxi-9α-tosilisocariolano 19, producto que posee en el C9 un buen grupo saliente. Este producto fue sometido a condiciones ácidas (HBr, 47%), obteniéndose como producto mayoritario el producto de reagrupamiento triciclo-[7.2.1.02,5]-1,4,4-trimetildodecan-8-ona 11, hecho que confirma nuestra hipótesis. Un minucioso estudio de la mezcla de reacción condujo al aislamiento de tosilato de metilo 20, cuya aparición puede explicarse mediante un mecanismo intramolecular (Fig. 10).

Dada la similitud estructural de este tipo de compuestos con los cariolanos, los cuales habían mostrado cierta actividad inhibitoria, se creyó oportuno someterlos a bioensayos para comprobar si poseían actividad frente a Botrytis cinerea. Concretamente fueron sometidos a estudio el isocariolan-9α-ol 16, el 8β-metoxi-isocariolan-9α-ol 8 y el isocariolan-8β,9α-diol 10.
Como puede observarse en los datos de inhibición (I) mostrados en las tablas 1-3 (2), el producto 8 podría considerarse dentro del grupo de compuestos con importante o elevada actividad, el 10 presentaría moderada actividad y el 16 poseería escasa o nula actividad. El 8β-metoxi-isocariolan-9α-ol 8 presenta el mayor porcentaje de inhibición [I (%)].



Puede observarse que el comportamiento de ambos productos 8 y 10 puede enmarcarse dentro del grupo de compuestos con actividad con un amplio rango de valores de I (%) dependiendo de la concentración a la que se ensaya, siendo especialmente activos a partir de 100 ppm y alcanzando a 200 ppm su mayor grado de inhibición. En un intento por establecer una relación estructura-actividad parece imprescindible que en la posición C8 se disponga de una función oxigenada. Así mientras que en el producto 16 en dicha posición la molécula dispone de un hidrógeno, en los compuestos 8 y 10 el sustituyente es un grupo metoxilo y un grupo hidroxilo, respectivamente. Estos resultados abren el camino a un estudio más profundo de este tipo de esqueletos y su potencial actividad frente a Botrytis cinerea. Sin lugar a dudas, un experimento de biotransformación de alguno de estos productos por el hongo, junto con estudios de acción-destoxificación, podría proporcionarnos datos para proponer un mecanismo de acción.
Parte Experimental
Métodos Generales. Los puntos de fusión están sin corregir. La TLC se llevó a cabo en gel de sílice 60 F254 (Merck) de 0.2 mm de espesor y con indicador fluorescente. Para la realización de la cromatografía en columna se usó gel de sílice SDS de grano 60-200 micrones compactada con gel seca o alúmina neutra Aldrich (150 mesh). Para la purificación por HPLC se empleó columnas de gel de sílice (Hibar 60, 7 ìm, de 1 x 25 cm de dimensiones). La preparación de las muestras se realizó con una pequeña columna de gel de sílice de 0.6 × 7 cm para eliminar la línea base y filtrado a través de filtros Teknokroma de 0.45 µm de poro. Los eluyentes utilizados fueron: hexano, acetato de etilo y mezcla de los mismos. Los disolventes se destilaron antes de su uso y además se filtraron sobre filtros Millipore de 0.45 µm de tamaño de poro.
Técnicas Microbiológicas. La cepa de Botrytis cinerea utilizada en este trabajo (UCA 992), se obtuvo de uvas de un viñedo de la empresa Allied & Domecq S.A. de España. Dicha cepa se encuentra depositada en el Departamento de Microbiología de la Universidad de Cádiz. Las cepas del antagonista Trichoderma harzianum (CECT 2413 y IMI 206040) fueron proporcionadas por la Colección Española de Cultivos Tipo (Valencia) y se encuentran depositadas en el Departamento de Genética de la Universidad de Sevilla. Como medio de cultivo se empleó agar-malta. Este medio de cultivo consta de 20 g de glucosa, 20 g de extracto de malta y 1 g de peptona, disueltos en un litro de agua destilada o desionizada. Seguidamente se ajusta la acidez del medio a pH 6.5-7, añadiéndose 20 g de agar.como sustrato de crecimiento de Botrytis cinerea. Tras ser esterilizado y dejado enfriar (45-50 ºC), se toman alícuotas de 20 mL, a las que se añaden volúmenes de 50 mL de etanol, conteniendo éstas concentraciones de 50, 100, 150 y 200 ppm del producto objeto del bioensayo. Cada concentración se probó por triplicado, de igual manera que los controles, para los que se añadieron 50 mL de etanol al medio, sin adicionar productos. Las mezclas líquidas de medio y producto se vierten, en condiciones de esterilidad, sobre placas Petri de 90 mm de diámetro. Una vez enfriadas éstas, se reemplaza de cada una de ellas un cilindro de 5 mm de diámetro por otro de igual dimensión y recubierto por cultivo de Botrytis cinerea.
Ciclación de óxido de cariofileno (1) catalizada por tetracianoetileno. A 200 mg de óxido de cariofileno 1 disueltos en 1 mL de metanol se le añaden 11.6 mg de tetracianoetileno y se mantiene la reacción con agitación durante 24 horas a temperatura ambiente. El progreso de la reacción se controla mediante cromatografía en capa fina (CCF). Una vez consumido el epóxido de partida se evapora el disolvente a presión reducida en un rotavapor. La mezcla de reacción obtenida se redisuelve en un disolvente más volátil (acetato de etilo) y se seca sobre Na2SO4 anhidro. La posterior evaporación del disolvente conduce a una mezcla de reacción que es purificada por columna cromatográfica, utilizando como relleno gel de sílice y mezclas de polaridad creciente de acetato de etilo en éter de petróleo. Los productos obtenidos son: 2β-metoxiclovan-9α-ol 4 (39%), 8β-metoxi-5α-hidroxicariofilan-3(4)-eno 5 (3%), isocariolan-9-ona 6 (11.5%) y una fracción compuesta por otros productos minoritarios 2-3 (30%).
Isocariolan-9-ona (6). Aceite. [α]25D -13.3º (c 2.93, CHCl3); IR (CHCl3) νmax 2952, 2865, 1704, 1456, 1367, 1111 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.88 (s, 3H, H-13), 0.88 (s, 3H, H-14), 0.96 (s, 3H, H-15), 1.16 (dddd, J =3.8, 5.6, 1.3, 1.3 Hz, H-7), 1.33 (ddt, J = 5.2, 13.1, 12.9 Hz, H-6), 1.48-1.43 (m, 1H, superpuesto con H-11, H-6), 1.47 (dd, 1H, J = 10.0, 10.3 Hz, H-11), 1.55 (dd, 1H, J = 10.2, 7.7 Hz, H-3), 1.72 (dd, 1H, J = 2.5, 14.0 Hz, H-12), 2.03 (dd, 1H, J = 9.6, 14.0 Hz, H-12), 2.27 (m,1H, H-7), 2.52-2.39 (m, 3H, H-8, H-10 y H-10'); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) δ 215.7 (C-9), 46.4 (C-5), 45.0 (C-8), 42.2 (C-2, C-11), 36.8 (C-3, C-12), 36.0 (C-10), 33.1 (C-4), 31.9 (C-1), 30.7 (C-15), 30.0 (C-7), 26.2 (C-13), 25.8 (C-6), 22.0 (C-14); EMIE m/z (int. rel.): 220 (26)[M]+, 205 (4) [M-OCH3]+, 177 (15) [M-CH3-CO]+, 169 (100), 146 (68).
Reacción del óxido de cariofileno con TCNE en acetona. Se disuelven 300 mg de óxido de cariofileno en 7.5 mL de acetona, añadiéndose posteriormente 600 mg de LiBr. Se añade una cantidad catalítica de TCNE (25 mg) y se agita la mezcla de reacción a lo largo de 4 horas, tras las cuales se evapora el disolvente a presión reducida. El sólido resultante se redisuelve en acetato de etilo y se seca sobre Na2SO4 anhidro. El disolvente es evaporado nuevamente obteniéndose una mezcla de reacción que al ser purificada por cromatografía en columna, empleando gel de sílice con gradientes crecientes de acetato de etilo en éter de petróleo, aislándose los compuestos siguientes: 1R,2S,5R,8S-8-carbaldehído-1,4,4-trimetiltriciclo[ 6.2.1.02,5 ]undecano 9 (5%), cariofila-3,8 (13)-dien-5α-ol 5 (20%), cariofila-4 (12)-8 (13)-dien-5α-ol 7 (70%).
Metanólisis de cariofila-4(12)-8(13)-dien-5α-ol (7) con TCNE. A 200 mg de cariofila-4 (12)-8 (13) dien-5α-ol 7 disueltos en 4 mL de metanol se le añaden 27 mg de TCNE y se mantiene la reacción a temperatura ambiente durante 48 horas. El progreso de la reacción se controla mediante cromatografía en capa fina. Al término de la misma se evapora el disolvente en un rotavapor y y se purifica por cromatografía en columna obteniéndose: isocariolan-9-ona 9 (15%), 8β-metoxi-isocariolan-9α-ol 8 (60%), 1R,2S,5R,8S-8-carbaldehído-1,4,4-trimetiltriciclo[ 6.2.1.02,5 ]undecano 9 (20%).
8β-metoxi-isocariolan-9α-ol (8). Aceite. [α]25D -12.12º (c 3.0, CHCl3,); IR (CHCl3) νmax 3381, 2923, 2336, 1728, 1704, 1655, 1471, 1082, 1013, 964 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.78 (s, 3H, H-14), 0.89-0.85 (d, 1H, J = 13.04 Hz, H-12), 0.94 (s, 3H, H-13); 0.95 (s, 3H, H-15), 1.23-1.18 (dd, 1 H, J = 5.16 Hz, H-11α), 1.28-1.24 (ta, 1H, H-3β), 1.56-1.48 (m,1H, H-3α), 1.76- 1.67 (m,3H, H-10α, H-11β, H-6β), 1.8-1.77 (m, 1H, H-10β), 1.86-1.83 (d, 1H, J = 13.04 Hz, H-12'), 1.9-1.84 (m, 1H, H-5β), 2.1-2.02 (m, 1H, H-2α), 3.21 (s, 3H, H-16), 3.56-3.52 (dd, J = 5.8, 11.23 Hz, 1H, H-9β); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) ä 215.7 (C-9), 46.4 (C-5), 45.0 (C-8), 42.2 (C-2, C-11), 36.8 (C-3, C-12), 36.0 (C-10), 33.1 (C-4), 31.9 (C-1), 30.7 (C-15), 30.0 (C-7), 26.2 (C-13), 25.8 (C-6), 22.0 (C-14); EMIE m/z (int. rel.): 275 [M + Na]+, 251 (4) [M-1]+, 221, 203, 193. EMAR: encontrado m/z: 275.1985 [M + Na]+; calcd para C16NaO2H28 m/z: 275.1987.
Reducción de 1R,2S,5R,8S-8-carbaldehído-1,4,4-trimetil-triciclo[6.2.1.02,5]undecano (9) con NaBH4. 700 mg de 1R,2S,5R,8S-8-carbaldehído-1,4,4-trimetiltriciclo[6.2.1.02,5] undecano 9 fueron disueltos en metanol y tratados con 200 mg de NaBH4 y mantenidos en agitación mecánica durante 24 horas. Al término de la misma, se añadió agua para destruir el exceso de NaBH4. Tras extracción con acetato de etilo y purificación en columna cromatográfica se obtuvo como producto mayoritario 1R,2S,5R, 8S-8-hidroximetil-1,4,4-trimetiltriciclo [6.2.1.02,5 ]undecano (89%).
Tratamiento de 1R,2S,5R,8S-8-hidroximetil-1,4,4-trimetil-triciclo[6.2.1.02,5]undecano con cloruro de dinitrobenzoilo. En 2mL de piridina se disuelven 25 mg de 1R,2S,5R,8S-8-hidroximetil-1,4,4-trimetiltriciclo[ 6.2.1.02,5 ]undecano y se tratan con 50 mg de cloruro de dinitrobenzoilo. Al cabo de 72 horas y tras comprobar el término de la reacción mediante CCF, se añade acetato de etilo y se lava con HCl 2N. Tras purificación mediante cromatografía en columna con gradiente creciente de acetato de etilo en éter de petróleo se obtiene: 1R,2S,5R,8S-8-(3,5,-dinitrobenzoilmetil)-1,4,4-trimetiltriciclo[6.2.1.02,5]undecano (85%).
1R,2S,5R,8S-8-(3,5,-Dinitrobenzoilmetil)-1,4,4-trimetiltriciclo[6.2.1.02,5]undecano. Sólido cristalino: pf 129-141 ºC; [α]25D -0.014 (c 2.6, CHCl3); IR (CHCl3) νmax 3099, 2954, 2860, 2332, 1734, 1659, 1627, 1522, 1464, 1345, 1275, 1162, 923, 722, 672 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.95 (s, 3H, H-12), 1.0 (s, 3H, H-14), 1.0 (s, 3H, H-13), 1.19-1.16 (d, 1H, J 11-11' = 12.4 Hz; H-11'), 1.41-1.37 (m, 3H, H-3, H-3', H-6'), 1.5-1.46 (m, 2H, H-9', H-7'), 1.54-1.52 (ta, 1H, J = 4.3 Hz; H-5), 1.61-1.56 (m, 1H, H-10), 1.66-1.63 (dd, 1H, J = 10.3 Hz; H-7), 1.71-1.68 (m, 2H, H-9, H-6), 1.83-1.73 (d, 1H, J = 12.1 Hz; H-11), 4.2 (s, 2H, H-15), 9.15-9.13 (dd, 2H, J = 0.9, 1.3 Hz; H-2', H-6'), 9.24-9.23 (m, 1H, H-4'); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) δ 148.7 (C-1'), 134.2 (C-3', C-5'), 129.3 (C-4'), 122.3 (C-2', C-6'), 77.1 (C-15), 48.9 (C-5), 48.2 (C-11), 45.9 (C-2), 45.8 (C-8), 45.2 (C-9), 41.5 (C-4), 37.2 (C-7), 34.5 (C-3), 33.5 (C-1), 30.5 (C-14), 28.9 (C-10), 25.6 (C-6), 22.3 (C-12), 20.4 (C-13); EMIE m/z (int. rel.): 416 [M]+, 360, 343, 195. EMAR: encontrado m/z: 416.1962 [M]+; calcd para C22O6H28N2 m/z: 416.1947 encontrado m/z: 360.1350[M-C4H8]+; calculado para C18O6H20N2 m/z: 360.1321. Estructura confirmada por análisis de difracción de rayos X.
Reacción de 8β-metoxi-isocariolan-9α-ol (8) con DEADcat /trifenilfosfina en presencia de CH2Cl2. Se disuelven 100 mg de 8β-metoxi-isocariolan-9α-ol 8 en la mínima cantidad de tolueno y se agitan magnéticamente. Se añaden 210 mg de trifenilfosfina y se les adiciona gota a gota 0.3 mL de DEAD y 20 mL de CH2Cl2, mantiéndolos a reflujo (<110 ºC) durante 4-5 h. Transcurrido este tiempo, se añade metanol y se evapora el disolvente a presión reducida. El crudo de reacción se purifica mediante columna cromatográfica eluyendo con polaridades crecientes de acetato de etilo y hexano, obteniéndose: 9β-cloro-8β-metoxi-isocariolano 14 (22%), 9α-cloro-8β-metoxi-isocariolano 15 (20%) junto con el producto de partida 8β-metoxi-isocariolan-9α-ol (8) (25%).
Reacción de 8β-metoxi-isocariolan-9α-ol (40) con DEADexc/trifenilfosfina en presencia de CH2Cl2. Se disuelven 100 mg de 8β-metoxi-isocariolan-9α-ol 8 en la mínima cantidad de tolueno y se agitaron magnéticamente. Se añaden 210 mg de trifenilfosfina y se les adiciona gota a gota 1 mL de DEAD y 20 mL de CH2Cl2, mantiéndolos a reflujo (<110º C) durante 4-5 h. Transcurrido este tiempo, se añade metanol y se evapora el disolvente a presión reducida. El crudo de reacción se purifica mediante columna cromatográfica obteniéndose: 9β-cloro-8β-metoxi-isocariolano 14 (18%), 9α-cloro-8β-metoxi-isocariolano 15 (15%), triciclo [ 7.2.1.02,5 ]-1,4,4-trimetildo-decan-8-ona 11 (4%), junto con el producto de partida 8β-metoxi-isocariolan-9α-ol 8 (20%).
Triciclo [7.2.1.02,5]-1,4,4-trimetildodecan-8-ona (11). Sólido cristalino: pf 129-137ºC; [α]25D +1.8 (c 2.4, CHCl3); IR (CHCl3) νmax 2951, 2934, 2860, 1736, 1461, 1381, 1123, 1104 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.86 (s, 3H, H-13α), 0.96 (s, 3H, H-15), 0.97 (s, 3H, H-14β), 1.41-1.36 (dd, 1H, J = 12.1 Hz, H-12α), 153-1.47 (m, 1H, H-7β), 1.55-1.53 (dd, 1H, H-11α), 1.83-1.66 (m, 2H, H-6β, H-11β), 2.00-1.87 (ddd, 1H, J = 2.9, 3.5 Hz, H-10β), 2.19-2.1 (m, 3H, H-2α, H-6α, H-10α), 2.3-2.23 (dd, 1H, J = 12.1 Hz, H-12β), 2.55-2.44 (ddd, 1H, H-9β), 2.87 -2.73 (dddd, 1H, H-7α); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) δ 219.3 (C-8), 50.9 (C-9), 45.8 (C-5), 41.8 (C-1), 41.1 (C-12), 40.1 (C-2), 38.4 (C-4), 38.1 (C-11), 34.3 (C-3), 31.4 (C-4), 30.6 (C-15), 29.4 (C-6), 25.5 (C-10), 23.7 (C-13), 23.5 (C-14); EMIE m/z (int. rel.): 220 [M]+, 201 [M-H2O]+, 187, 164, 148, 135, 121. EMAR: encontrado m/z: 220.1839 [M]+; calculado para C15O H24: 220.1827. Estructura confirmada por análisis de Difracción de RX de un derivado de reducción.
9β-Cloro-8β-metoxi-isocariolano (14). Sólido cristalino: pf 119-125ºC; [α]25D +1.8 (c 2.1, CHCl3); IR (CHCl3) ímax 3266, 2947, 2634, 2378, 1706, 1503, 1457, 1382, 1097 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.84 (s, 3H, H-13α), 0.97 (s, 3H, H-15), 0.98 (s, 3H, H-14β), 1.34-1.26 (m, 1H, H-3β), 1.58-1.44 (m, 3H, H-3α, H-10, H-11β), 1.84-1.72 (m, 3H, H-11α, H-6β, H-5β), 2.12-2.02 (m, 1H, H-10'), 2.34-2.26 (m, 1H, H-10), 3.22 (s, 3H, H-16), 4.19 (sa, 1H, H-9α); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) δ 78.5 (C-8), 63.4 (C-9), 48.1 (C-16), 43.9 (C-5), 40.5 (C-12), 37.2 (C-2), 35.4 (C-3), 34.8 (C-4), 32.5 (C-1), 31.9 (C-11), 30.6 (C-15), 29.9 (C-7), 29.3 (C-10), 26.2 (C-13), 20.9 (C-6), 20.8 (C-14); EMIE m/z (int. rel.): 270 [M]+, 255 [M-15]+, 235, 205, 203, 194, 193, 187, 183, 179, 174. EMAR: encontrado m/z: 235.2093 [M-Cl]+; calculado para C16OClH27: 235.2062. Estructura confirmada por análisis de difracción de rayos X.
9α-Cloro-8β-metoxi-isocariolano (15). Sólido: pf 111-119ºC; [α]25D -1.2 (c 1.3, CDCl3,); IR (CHCl3) νmax 2947, 2936, 2856, 1730, 1381, 1444, 1077, 852, 550 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.84 (s, 3H, H-13α), 0.97 (s, 3H, H-15), 0.98 (s, 3H, H-14β), 1.65- 1.62 (m, 1H, H-3β), 1.83-1.66 (m, 3H, H-3α, H-10α, H-11α), 2.13-2.01 (m, 3H, H-5β, H-11β, H- 6β), 2.39-2.3 (m, 3H, H-12, H-2α, H-10β), 3.21 (s, 3H, H-16), 4.37-4.35 (dd, 1H, J = 5.4, 3.4 Hz, H-9β); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HECTOR) δ 78.0 (C-8), 59.5 (C-9), 48.1 (C-16), 43.9 (C-5), 41.2 (C-12), 37.6 (C-2), 35.5 (C-3), 34.8 (C-4), 32.5 (C-1), 32.8 (C-11), 30.6 (C-15), 30.1 (C-7), 29.0 (C-10), 26.2 (C-13), 21.1 (C-6), 20.8 (C-14); EMIE m/z (int.rel.): 235 [M-Cl]+, 193, 180, 151, 137. EMAR: encontrado m/z: 235. 2048 [M-Cl]+; calculado para C16OClH27 : 235.206.
Reacción de isocariolan-9α-ol (20) con DEAD/trifenilfosfina. En la mínima cantidad de tolueno se disuelven 42 mg de isocariolan-9α-ol 16, a los que se le añaden 150 mg de PO3 y 0.5 ml de DEAD, manteniéndose la mezcla de reacción bajo reflujo (< 110 ºC) durante 7 h. Al término de las mismas se añade MeOH para evaporar con facilidad el tolueno residual y se purifica el crudo mediante cromatografia en columna y HPLC obteniéndose: isocariolan-9-dietoxicarbonilhidrazina 17 (52%) y producto de partida 16 (40%).
Isocariolan-9-dietoxicarbonilhidrazina (17). Aceite; [α]25D - 0.8 (c 1.2, CHCl3); IR (CHCl3) νmax 3014, 2915, 2760, 2214, 1925, 1625, 1414, 1322, 1129, 921, 845 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.79 (s, 3H, H-15), 0.98 (s, 3H, H-14β), 0.98 (s, 3H, H-13α), 1.2- 1.12 (dd, 1H, H-12'), 1.47-1.42 (t, 6H, H-18, H-21), 1.8-1.76 (m, 2H, H-10α, H-12), 1.84-1.8 (m, 1H, H-10β), 2.12-2.04 (m, 1H, H-2α), 2.46 (sa, 1H, H-8β), 4.52-4.46 (dd, 4H, J = 7.1, 14.3 Hz; H-17, H-17', H-20, H-20'), 5.08-5.02 (m, 1H, J = 5.8, 6.2 Hz; H-9α); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) δ 160.4 (C-19), 159.9 (C-16), 82.1 (C-9), 65.4 (C-17, C-20), 46.2 (C-5), 39.8 (C-12), 39.6 (C-8), 38.1 (C-11), 36.1 (C-3), 35.3 (C-2), 34.4 (C-4), 31.4 (C-1), 30.6 (C-15), 26.2 (C-10), 24.9 (C-13), 24.2 (C-7), 24.0 (C-6), 20.6 (C-14), 14.1 (C-18, C-21); EMIE m/z (int. rel.): 380, 302, 226, 220, 205, 204, 198, 190, 176.
Reacción de isocariolan-9α-ol (16) con DEAD/trifenilfosfina en CH2Cl2. En la mínima cantidad de tolueno se disuelven 50 mg de isocariolan-9α-ol 16, a los que se le añaden 150 mg de PÖ3, 0.5 mL de DEAD y 12 mL de CH2Cl2, manteniéndose la mezcla de reacción bajo reflujo (< 110 ºC) durante 7 horas. Posteriormente se añade MeOH para evaporar con facilidad el tolueno residual y tras purificar el crudo mediante cromatografia en columna y HPLC se obtiene: 9β-cloro-isocariolano 18 (45%) y producto de partida 16 (50%).
9β-Cloro-isocariolano (18). Aceite; [α]25D - 0.8 (c 1.2, CHCl3); IR (CHCl3) νmax 3129, 2984, 2654, 2104, 1615, 1525, 1484, 1124, 976, 724 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.76 (s, 3H, H-15), 0.97 (s, 3H, H-14β), 0.98 (s, 3H, H-13α), 1.15-1.1 (dd, 1H, J = 4.7 Hz, H-12β), 1.73-1.63 (m, 2H, H-12α, H-10β), 2.01-1.9 (ddd, 1H, J = 4.2, 4.4 Hz, H-10α), 2.13-2.05 (ddd, 1H, J = 8.0, 7.8, 3.4 Hz, H-2α), 2.29 (sa, 1H, H-8β), 4.96-4.9 (ddd, 1H, J = 11.7, 5.7, 6.0 Hz, H-9β); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) δ 75.7 (C-9), 46.4 (C-5), 39.9 (C-12), 39.8 (C-8), 38.2 (C-11), 36.1 (C-3), 35.4 (C-2), 34.3 (C-4), 31.4 (C-1), 30.6 (C-15), 26.4 (C-13), 25.3 (C-7), 24,4 (C-10), 24.3 (C-6), 20.6 (C-14); EMIE m/z (int.rel.): 240, 239, 205, 203, 194, 193, 190, 179, 174.
Reacción de tosilación de 8β-metoxi-isocariolan-9α-ol (8). A 300 mg de 8β-metoxi-isocariolan-9α-ol 8 se le añaden 50 mg de cloruro de tosilo disueltos en piridina y se mantienen a temperatura ambiente y con agitación durante siete días. La reacción se controla mediante CCF. Al término de la reacción se añade acetato de etilo. El crudo de reacción obtenido se lava dos veces con HCl 2N y con NaClsat, posteriormente se seca sobre Na2SO4 anhidro y tras su filtración se evapora el disolvente a presión reducida, obteniéndose como producto mayoritario 8β-metoxi-9α-tosil-isocariolano 19 (90%).
8β-Metoxi-9α-tosil-isocariolano (19). Aceite; [α]25D - 2.3 (c 3.6, CHCl3); IR (CHCl3) νmax 3024, 2925, 2715, 2314, 1944, 1601, 1543, 1351, 1145, 980, 675 cm-1; RMN 1H (CDCl3, 400 MHz) δ 0.77 (s, 3H, H-13α), 0.94 (s, 3H, H-15), 0.95 (s, 3H, H-14β), 0.98- 0.92 (d, 1H, J = 13.2 Hz, H-12), 1.77-1.69 (dd, 1H, J = 2.9, 13.2 Hz, H-12'), 2.08-1.98 (m, 1H, H-10α), 2.41 (s, 3H, H-7'), 2.86 (s, 3H, H-16), 4.5-4.47 (t, 1H, J = 8.6 Hz, H-9β), 7.3-7.26 (m, 2H, H-3', H-5'), 7.8-7.76 (d, 2H, J = 8.4 Hz; H-2', H-6'); RMN 13C (CDCl3, 50 MHz, asignaciones por APT y HETCOR) δ 170.4 (C-16), 81.4 (C-9), 75.1 (C-8), 48.5 (C-12), 45.4 (C-5), 38.3 (C-2), 37.2 (C-11), 35.9 (C-7), 34.8 (C-4), 34.1 (C-3), 32.7 (C-1), 30.6 (C-15), 26.2 (C-10), 25.9 (C-17), 21.3 (C-13), 21.1 (C-6), 20.7 (C-14); EMIE m/z (int.rel.): 406, 252, 251, 220, 202, 192, 190, 174.
Reagrupamiento de 8β-metoxi-9α-tosil-isocariolano (19) con HBr. 38 mg de 8β-metoxi-9α-tosil-isocariolano 19 disueltos en 12 mL de acetona se le añaden 12 mL de HBr (47%), manteniendo la mezcla en agitación durante 24 horas. Tras la extracción con acetato de etilo y purificación de la mezcla de reacción por cromatografía en columna se obtiene: triciclo [7.2.1.02,5]-1,4,4-trimetildodecan-8-ona 11 (60%) y tosilato de metilo 20 (6%).
Referencias
1. Nikon, A. J. Amer. Chem. Soc. 1955, 77, 1190-1196. [ Links ]
2. Tkachev, A. V.; Gatilov, Y. V.; Bagryanskaya, I. Y. Zh. Org. Khim. 1985, 490-503. [ Links ]
3. Tkachev, A. V.; Mamatyuk, V. I.; Dubovenko, Zh. V. Zh. Org. Khim. 1991, 1469-1475. [ Links ]
4. Khomenko, T.M.; Korchagina, D. V.; Gatilov, Y. V. Zh. Org. Khim. 1991, 1839-1852. [ Links ]
5. Rebordinos, L; Cantoral, J.M; Hanson, J.R.; Collado, I.G. Phytochemistry. 1996, 42, 383. [ Links ] Phytochemistry 1996, 41, 513. [ Links ]
6. Merrifield, R. E.; Phillips, W. D. J. Amer. Chem. Soc. 1958, 80, 2778-2782. [ Links ]
7. Masaki, Y.; Ochiai, M. Chem. Lett. 1993, 17-20. [ Links ]
8. Masaki, Y.; Ochiai,. Synlett. 1993, 847-849. [ Links ]
9. Masaki, Y.; Ochiai, Bull. Chem. Soc. Jpn. 1996, 69, 195-205. [ Links ]
10. Masaki, Y.; Ochiai,. J. Chem. Soc. Perkin Trans I. 1994, 1659-1660. [ Links ]
11. Tanemura, K.; Suzuki, T.; Horaguchi, T. Bull. Chem. Soc. Jpn. 1994, 67, 290-292. [ Links ]
12. Macías, A.; Collado, I. G.; Hanson, J. R. J. Org. Chem. 1997, 62, 1965. [ Links ]
13. Macías, A.; Collado, I. G.; Hanson, J. R. Tetrahedron 1996, 52, 7961-7982. [ Links ]
14. Collado, I. G.; Hanson, J. R.; Racero, J.C. Tetrahedron 1998, 54, 1615-1626. [ Links ]
15. Los datos cristalográficos han sido depositados en el Centro de Datos Cristalográficos de Cambridge, pudiendo ser obtenidos a través de: Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK.
16. Racero, J. C.; Hanson, J. R.; Collado, I. G. J. Org. Chem. 2000, 65, 7786-7791. [ Links ]
17. Mitsunobu, O. Synthesis 1981, 1-28. [ Links ]
18. Collado, I. G.; Hanson, J. R.; Racero, J. C. Tetrahedron Lett. 1999, 40, 6497-6498. [ Links ]
19. Loibner, H.; Zbiral, E. Helv. Chim. Acta. 1976, 59, 2100. [ Links ]

