










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 no.3 Ciudad de México jul./sep. 2003
Investigación
Síntesis de nuevos monómeros para fotopolimerizaciones catiónicas
Ricardo Acosta Ortiz,1* y James V. Crivello2
1 Centro de Investigación en Química Aplicada, Boulevard Enrique Reyna #140, C.P.25140, Saltillo, Coahuila, México. Tel. (844) 438-9830; Fax (844) 4 38 98 39. E-mail racosta@polimex.ciqa.mx
2 Rensselaer Polytechnic Institute, 101 8th Street, Troy, New York, 12180, USA.
Recibido el 23 de abril del 2003
Aceptado el 24 de septiembre del 2003
Resumen
Se reportan los métodos de preparación de cinco nuevos monómeros susceptibles de ser fotopolimerizados catiónicamente. Estos incluyen compuestos del tipo epoxiciclohexano, vinil éter, propenil éter y oxetanos. La característica común de estos monómeros es que contienen en su estructura un grupo bencílico con sustituyentes donadores de electrones como los grupos metóxido o el grupo metilendioxolano. La síntesis de estos compuestos involucra reacciones de eterificación y oxidación. Se explica el razonamiento para el diseño de los monómeros.
Palabras clave: Monómeros, fotopolimerización, catiónica, radical, síntesis.
Abstract
The synthesis of five novel cationically photopolymerizable monomers is reported. Vinyl ether, propenyl ether, oxetanes and epoxycyclohexane moieties are included. All of these compounds bear benzyl ether group with electron donor groups, such as methoxy or methylene dioxolane groups. The preparation of these compounds involves etherification and oxidation reactions. The rationale behind the design of these monomers is explained.
Keywords: Monomers, photopolymerization, cationic, radicals, synthesis.
Introducción
Las polimerizaciones fotoiniciadas ya sean de tipo radical o catiónico han encontrado una amplia aplicación en las áreas de recubrimientos, tintas de impresión, adhesivos, circuitos electrónicos, holografía y aplicaciones dentales [1-3]. Entre las ventajas de las fotopolimerizaciones se puede destacar las altas velocidades de polimerización, lo que resulta en un polímero completamente curado en el intervalo de segundos. Esto permite que las fotopolimerizaciones puedan implementarse exitosamente a nivel industrial. En este proceso se hacen pasar rápidamente las piezas a recubrir, mediante una banda transportadora a través de una unidad de lámparas UV de alta intensidad y se logra curar el polímero totalmente al pasar por dicha unidad.
Debido a lo anterior, es importante resaltar que la reactividad del sistema a fotocurar es de vital importancia para su factibilidad en líneas de producción. Un sistema a ser fotopolimerizado o fotocurado consiste de un monómero usado como diluyente y modificador de viscosidad, uno o más oligómeros que dan al material sus características poliméricas y un fotoiniciador. La fotopolimerización se origina al irradiar la muestra con luz UV o haces de electrones.
Tradicionalmente las fotopolimerizaciones más comunes se dividen en dos grupos: a) las fotopolimerizaciones radicálicas que usan monómeros vinílicos o acrílicos, y b) las fotopolimerizaciones catiónicas en las cuales se usan monómeros epóxicos, éteres vinílicos y algunos heterociclos como derivados de tetrahidrofurano y oxetanos. Las excelentes propiedades químicas y mecánicas obtenidas de los polímeros derivados de estos últimos monómeros [4], aunado a la insensibilidad al oxígeno, han suscitado un gran interés para el desarrollo de sistemas catiónicos fotocurables cada vez más reactivos.
El descubrimiento en los años setenta [5, 6], de que algunas sales de onio como las sales de diariliodonio y triarilsulfonio mostraban alta reactividad como fotoiniciadores de monómeros polimerizables catiónicamente, fue un factor determinante para el rápido desarrollo de esta área de las fotopolimerizaciones catiónicas, tanto en el aspecto científico como en el tecnológico
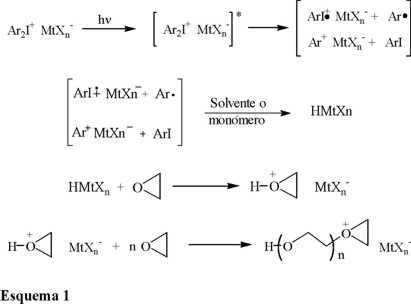
En el esquema 1 se presenta el mecanismo de iniciación de las fotopolimerizaciones catiónicas mediante el uso de sales de diariliodonio [7]. La fotólisis de estas sales resulta en la formación de radicales, cationes y catión-radicales (Ec. 1). Las dos especies catiónicas interaccionan con el monómero o impurezas para generar ácidos protónicos (Ec. 2). Estos ácidos próticos son las especies primarias responsables de iniciar la polimerización (Ec. 3 y 4). Cabe hacer notar que solo el proceso de iniciación es fotoquímico. Otras sales de onio, como las de triarilsulfonio y trarilselenonio, muestran un mecanismo similar. También es importante resaltar en este esquema, la formación de radicales libres de tipo arilo, ya que estas especies participan en reacciones laterales que pueden influir en el curso de la polimerización.
Ledwith y Yagci [8,9] descubrieron que los radicales generados en la fotólisis de la 2-fenil, 2,2-dietoxiacetofenona eran capaces de reaccionar por medio de una reacción redox con las sales de diariliodonio reduciéndose éstas en el proceso y generando el carbocatión dietoxifenil, el cual fue capaz de iniciar una polimerización catiónica. De igual manera, Bi y Neckers [10] demostraron que los radicales producidos al extraer un átomo de hidrógeno al nitrógeno de aminas terciarias por la acción de un tinte fotoexcitado, también eran capaces de inducir la descomposición de las sales de diariliodonio.
En 1993 Crivello y colaboradores [11], descubrieron que las fotopolimerizaciones catiónicas de vinil éteres con grupos alilo, fotoiniciadas por sales de diariliodonio, mostraron un efecto de aceleración en comparación con las polimerizaciones en las cuales los monómeros no contenían el grupo alilo. Se demostró que este efecto podía adscribirse a la descomposición de las sales de diariliodonio por efecto de radicales libres. En el esquema 2 se muestra el mecanismo propuesto.
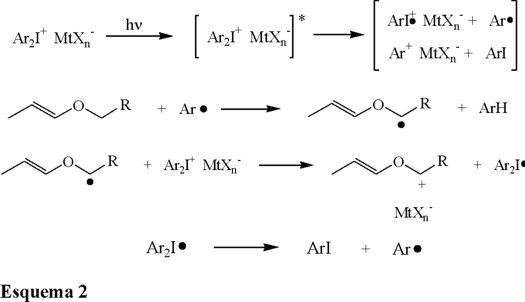
Los radicales arilo generados en la fotólisis de la sal de onio (Ec. 5) pueden extraer los átomos de hidrógeno al átomo de oxígeno del éter (Ec. 6). Esto produce un radical en la estructura del monómero, el cual es posteriormente oxidado por la sal de diariliodonio para formar un ion alcoxicarbenio (Ec. 7). Esta última especie es capaz de iniciar una polimerización. Al mismo tiempo, el radical diariliodo generado sufre una reacción irreversible de descomposición para dar el correspondiente radical arilo y iodobenzeno (Ec. 8). En conjunto, las ecuaciones 5-8 representan un proceso de reacción en cadena que resulta en la formación de una mayor cantidad de especies catiónicas, y esto se traduce en la aceleración del proceso total de polimerización.
Este concepto de nuevos monómeros conteniendo grupos lábiles que pueden interactuar con el fotoiniciador, ha dado lugar al diseño y síntesis de nuevos monómeros altamente reactivos para fotopolimerizaciones catiónicas [12-14]. Estudios recientes [15] han mostrado que en el caso de los bencil 3,4 epoxiciclohexilmetil éteres, la reactividad de estos monómeros se incrementa al introducir grupos donadores de electrones en el anillo aromático, cuya función es estabilizar tanto al radical como al carbocatión generado en la estructura del monómero.
En este artículo se presentan las síntesis de varios monómeros, cuya característica principal es la presencia de grupos bencílicos con sustituyentes de tipo metóxido, por lo que se espera que presenten altas velocidades de polimerización. En especial se han diseñado monómeros derivados del alcohol piperonílico, el cual es un producto natural de bajo costo que cumple con el requisito de un anillo con una alta densidad electrónica, producto del grupo sustituyente metilendioxolano, así como del alcohol 3,4,5 trimetoxibencílico.
Parte Experimental
Materiales y Equipo
Los reactivos utilizados en la síntesis de los monómeros fueron comprados en Aldrich y se usaron como se recibieron. Los espectros de RMN 1H y 13C fueron determinados en un equipo Gemini 2000 de 200 MHz Varian, usando CDCl3 como disolvente y Tetrametil silano como referencia, mientras que los espectros de IR se determinaron en un Magna 550 de Nicolet, y el análisis elemental en un equipo Perkin Elmer CHNS Analyzer serie 2400.
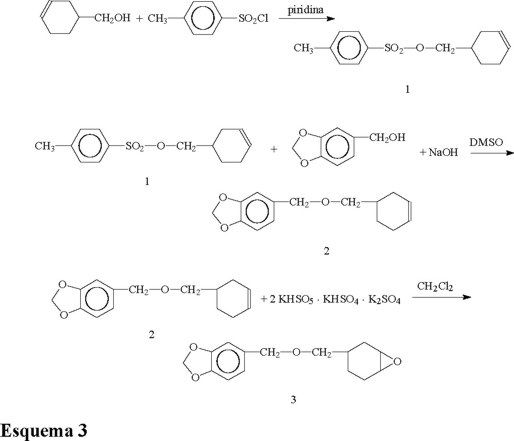
a) p-Toluensulfonato de ciclohexil-3-enilmetilo (1). En un matraz de 3 bocas de 100 mL provisto de agitación magnética, entrada para argón y termómetro, se disolvieron 20.42 g (0.107 mol) de cloruro de p-toluensulfonilo en 50 mL de piridina seca (BaO). Enseguida se agregaron lentamente 10 g (0.089 mol) del alcohol 1,2,3,6-tetrahidrobencílico. La mezcla de reacción se enfrió a 0 °C y se dejo 8 h en agitación manteniendo la temperatura. Al término de este tiempo, se dejó estabilizar la mezcla a temperatura ambiente y después se vertió esta mezcla en 200 mL de agua destilada. Se separaron las fases, la fase orgánica se redisolvió en diclorometano y se extrajo con 3 porciones de 50 mL de HCl al 10 %. Después se lavó con 3 porciones de una solución de cloruro de sodio saturada y posteriormente la fase orgánica se secó con sulfato de sodio anhidro. Al evaporar el solvente se obtuvieron 20.2 g de un aceite incoloro 1 (85 %). Debido a la inestabilidad térmica de los tosilatos no se procedió a purificar el compuesto por destilación y se purificó en su lugar por cromatografía en columna usando sílica gel como soporte y diclorometano como eluente.1: RMN 1H (CDCl3, 200 MHz) δ: 1.05-2.15 (7H, m, H cicloalifáticos), 2.41 (3H, s, CH3-Ar); 3.90 (2H, d, O-CH2-); 5.72 (2H, s, CH=CH-), 7.35-7.80 (4H, dd, Ar-H);. IR (KBr) νmax 3065, 3032, 2920, 2844, 1657, 1603, 1361, 1173, 1093, 964, 840, 819, 674, 561 cm−1.
b) (3,4-metilendioxi)bencil, ciclohex-3-enil-metil éter (2). En una matraz de tres bocas de 250 mL provisto de termómetro, condensador y agitación magnética se agregaron 29.3 g (0.110 mol) de 1, 18.43 g (0.121 mol) de alcohol piperonílico y 4.84 g (0.121 mol) de hidróxido de sodio en 100 mL de dimetil sulfóxido. La mezcla se calentó a 50 °C por 6 h y al cabo de este tiempo se dejó alcanzar la temperatura ambiente y se vertió en 200 mL de agua destilada. Se agregó entonces 100 mL de diclorometano y se separaron las fases. La fase orgánica se lavó continuamente con agua hasta eliminar por completo el sulfóxido de dimetilo. Enseguida se secó la fase orgánica con sulfato de sodio anhidro, posteriormente se filtró y se evaporó el solvente para obtener un líquido incoloro. Este líquido se purificó por medio de cromatografía en columna de tipo flash usando sílica como soporte y una mezcla de hexano-acetato de etilo 95:5 como eluente. Después se destiló a vacío obteniéndose un líquido incoloro a 120-122 °C a 8 mmHg. Se obtuvieron 22.2 g (82 %) del producto 2: Líquido incoloro p.eb. 120-122 °C a 8 mmHg. RMN 1H (CDCl3, 200 MHz) δ 1.19-2.15 (7H, m, H cicloalifáticos), 3.30 (2H, d, O-CH2-); 4.40 (2H, s, Ar-CH2); 5.65 (2H, s, -CH=CH-); 5.95 (2H, s, O-CH2-O); 6.81-6.85 (3H, m, Ar-H), IR (KBr) νmax: 3027, 2920,1651, 1608, 1254, 1098, 1039, 931, 808, 667 cm−1.
c). (3,4-metilendioxi)bencil, 3,4-epoxiciclohexilmetil éter (3). En un matraz de 2 L de tres bocas, equipado con termómetro, agitación magnética, y dos embudos de adición se agregaron 22.5 g (0.0914 mol) de 2 disueltos en 100 mL de acetona y 100 mL de diclorometano. Después se adicionaron 300 mL de una solución buffer (HydrionMR, pH 7.4) y 0.5 g de éter 18-Corona-6 como catalizador de transferencia de fase. La mezcla se enfrió a 0 °C. Posteriormente se empezó la adición lenta de 84.38 g (0.1371 mol) de Oxone (peroximono-sulfato de potasio, 2KHSO5.KHSO4.K2SO4) como una solución 1 N en agua destilada. Al mismo tiempo se agregó una solución 1 N de NaOH para mantener el pH neutro. Después de finalizar la adición del oxidante, la mezcla se continuó agitando por espacio de 12 h. Al cabo de este tiempo se separaron las fases y la fase orgánica se lavó continuamente hasta eliminar el catalizador de transferencia de fase y secó con sulfato de sodio anhidro. El solvente se evaporó y el residuo se destiló a vacío obteniéndose 17.25 g (72 %) de un líquido incoloro 3. 3: Líquido incoloro p.eb. 150-153 °C a 10 mmHg. RMN 1H (CDCl3, 200 MHz) δ 0.80-2.20 (7H, m, H ciclo alifáticos); 3.00 -3.10 (2H, m, H-C-O); 3.10-3.30 (2H, m, O-CH2-); 4.40 (2H, s, Ar-CH2-); 5.95 (2H, s, O-CH2-O); 6.80-6.85 (3H, m, Ar-H); IR (KBr): 3070, 2984, 1608, 1243, 1103, 1050, 931, 819, 556 cm−1; Anál : C 68.4 %, H 6.8 % calcd para C 68.7 %, H 6.9 %.
d) 3,4,5-Trimetoxibencil, 3,4-epoxiciclohexil-metil éter (4). El compuesto 4 derivado del alcohol 3,4,5-trimetoxibencílico se sintetizó utilizando la misma metodología usada para preparar el compuesto 3. Este se obtuvo en un rendimiento del 68 %. 4: Líquido ligeramente naranja, dec. 200 °C a 10 mmHg. RMN 1H (CDCl3, 200 MHz) δ: 0.90-2.30 (7H, m H cicloalifáticos); 3.10-3.20 (2H, m, H-C-O); 3.20-3.30 (2H, m, OCH2-); 3.80 (3H, s, p-Ar-O-CH3); 3.85 (6H, s, m-Ar-OCH3); 4.40 (2H, s, Ar-CH2-); 6.50 (2H, s, Ar-H); IR (KBr) max: 3070, 2941, 1731, 1586, 1232, 1113, 829, 786, 749, 695 cm−1 Anál. C 65.8 %, H 7.7 %, calcd para C 66.2 %, H 7.8 %.
e) (3,4-Metilendioxi)bencil, alil éter (5). En un matraz de 100 mL de tres bocas provisto de condensador, termómetro y agitación magnética se agregaron 15.21 g (0.1 mol) de alcohol piperonílico, 4 g (0.1 mol) de NaOH, 11 g (0.09 mol) de bromuro de alilo y 50 mL de tolueno. Esta mezcla se agitó por 15 min, al cabo de los cuales se agregaron 2.93 g (0.009 mol) de bromuro de tetrabutil amonio como catalizador de transferencia de fase. Lentamente se llevó la mezcla a temperatura de ebullición y se mantuvo así por 10 h. Después de este tiempo la mezcla se enfrió y se agregó a 200 mL de agua destilada. Se separaron las fases y la fase orgánica se lavó con tres porciones de 50 mL de agua destilada. Posteriormente la mezcla se secó con sulfato de sodio anhidro. Después de filtrar y evaporar el solvente, el residuo se destiló a vacío obteniéndose 16.04 g (91.8 %) de 5. 5: Líquido incoloro p.eb. 122-125 °C a 5 mmHg. RMN 1H (CDCl3, 200 MHz) δ: 4.00 (2H, d, CH2-CH=CH2); 4.40 (2H, s, Ar-CH2-); 5.20 (2H, m, -CH=CH2); 5.85 (1H, m, -CH=CH2); 5.95 (2H, s, O-CH2-O); 6.80-6.85 (3H, m, Ar-H); IR (KBr) νmax: 3075, 2995, 1641, 1614, 1512, 1243, 1162, 1039, 996, 910, 813, 765, 636, 523 cm−1.
f) (3,4-Metilendioxi)bencil, 1-propenil éter (6). En un matraz de 100 mL con termómetro, entrada para argón y agitación magnética se adicionaron 12 g del compuesto 5 y 5 mg del catalizador [(P[Ph]3)3RuCl2]. La temperatura se elevó entonces a 120 °C y se mantuvo por espacio de 4 h. Todo el proceso se llevó a cabo en atmósfera de argón. El proceso fue cuantitativo ya que al analizar por RMN de protón desaparecen completamente las señales alílicas y son reemplazadas por las de la mezcla de isómeros cis y trans del propenil éter correspondiente 6. Esta mezcla fue destilada a vacío obteniéndose 10 g de un líquido incoloro. 6: Líquido incoloro p.eb. 125-128 °C a 5 mmHg. RMN 1H (CDCl3, 200 MHz) δ: 1.55 (3H, dd, J = 1.53 Hz, CH3-CH=CH, isómero trans), 1.65 (3H, dd, J=1.83 Hz, CH3-CH=CH, isómero cis), 4.40 (1H, m, CH3-CH=CH, isómero cis), 4.60 (2H, s, Ar-CH2- isómero trans); 4.70 (2H, s, Ar-CH2-, isómero cis), 4.90 (1H, m, CH3-CH=CH-, isómero trans); 6.00 (2H, s, -O-CH2-O-); 6.10 (1H, m, CH3-CH=CH-, isómero cis); 6.30 (1H, m, CH3-CH=CH-, isómero trans); 6.80-6.85 (3H, m, Ar-H); IR (KBr) νmax: 3075, 3011, 2882, 2774, 1673, 1501, 1453, 1248, 1130, 1087, 1034, 937, 813 cm−1. Anal. Experim. C 74.3 %, H 6.4 %, calcd. para C 74.1 %, H 6.2 %.
g) 2-[(3,4-Metilendioxi)benciloxi]etil, vinil éter (7). En un matraz de 100 mL de tres bocas provisto de condensador, termómetro y agitación magnética se agregaron 15.70 g (0.1032 mol) de alcohol piperonílico, 4.13 g (0.1032 mol) de NaOH, 10 g (0.0938 mol) de 2-cloroetil vinil éter en 50 mL de DMSO y se calentó la mezcla de reacción a 50 °C por espacio de 10 h. Después de finalizar este tiempo, se vertió la mezcla en 200 mL de agua destilada y se agregaron 100 mL de diclorometano. Enseguida se separaron las fases y la orgánica se lavó con agua destilada hasta eliminar completamente el DMSO. La fase orgánica se secó con sulfato de sodio anhidro y se evaporó para obtener un líquido incoloro que se purificó por cromatografía en columna usando silica gel como soporte y una mezcla de hexano: acetato de etilo 95:5 como eluente. Se obtuvieron 15.8 g del compuesto 7 (68.8 %). 7: Líquido incoloro p.eb. 158-161 °C a 12 mmHg. RMN 1H (CDCl3, 200 MHz) δ: 3.65 (2H, m, Bz-O-CH2-CH2-O); 3.85 (2H, m, Bz-O-CH2-CH2-O-); 3.9 (1H, dd, J = 4.88 Hz; -CH = CH2); 4.2 (1H, dd, J =12.52 Hz; -CH=CH2); 4.45 (2H, s, Ar-CH2-O-); 5.9 (2H, s, -O-CH2-O-); 6.5 (1H, dd, J = 6.7 Hz; -CH=CH2); 6.80-6.85 (3H, m, Ar-H); IR (KBr) νmax: 3118, 3075, 2866, 1625, 1254, 1189, 1098, 1039, 931, 819 cm−1; Anal. C 65.1 %, H 6.4 %, calcd para C 64.9 %, H 6.3 %.
h) 3-Etil-3-hidroximetiloxetano (8). En un matraz de 250 mL de tres bocas provisto de termómetro, condensador y una cabeza de destilación se agregaron 67 g (0.5 mol) de trimetilol propano [2-etil-2-(hidroximetil)-1,3-propanodiol], 59 g (0.5 mol) de carbonato de dietilo y 0.05 g de KOH disuelto en 2 mL de alcohol absoluto. La mezcla se mantuvo en reflujo hasta que la temperatura de la mezcla de reacción alcanza 105 °C, y entonces el etanol generado se removió por destilación observándose una temperatura en la cabeza de destilación de 76-78 °C. La destilación se continuó hasta que la temperatura de la mezcla de reacción alcanzó los 145 °C. Entonces se aplicó vacío al sistema lentamente hasta llegar a 15 mmHg, manteniendo la temperatura de la mezcla a 140-150 °C. Entonces se elevó la temperatura a 180 °C y se observó el desprendimiento de dióxido de carbono y la mayoría del producto destiló a una temperatura entre 100-160 °C. El producto obtenido se somete nuevamente a una destilación a vacío obteniéndose 44 g (76 %) del compuesto puro 8. 8: Líquido incoloro p.eb. 114-115 °C a 16 mmHg. RMN 1H (CDCl3, 200 MHz) δ: 0.9 (3H, t, CH3-); 1.7 (2H, c, -CH2-); 3.25 (1H, t, -OH); 3.65 (2H, d, CH2-O); 4.4 (4H, dd, -CH2-O-CH2-); IR (KBr) νmax: 3403, 2968, 2882, 1652, 1458, 1044, 974, 776 cm−1.
i) Cloruro de 3,4,5-Trimetoxibencilo (9). En un reactor tubular de vidrio con alineación vertical, provisto de condensador, termómetro, salida de gases y entrada de gas por la parte inferior, se agregaron 100 mL de éter y 10 g de alcohol 3,4,5-trimetoxibencílico. El reactor se colocó en un baño de hielo para alcanzar una temperatura de 0 a 5 °C. Entonces se hizo pasar a través de este reactor, una corriente de cloruro de hidrógeno seco. Este gas se desprende al adicionar lentamente ácido clorhídrico concentrado en 100 mL de ácido sulfúrico concentrado en un matraz de 250 mL de tres bocas con salida para gases. Este matraz se conectó por medio de una manguera de teflón al reactor tubular. El gas pasó a través del seno de la solución y el gas que no reaccionó se hizo pasar a una trampa con agua destilada. Después de agregar 80 mL de ácido clorhídrico concentrado, se detuvo la reacción, se vertió en un vaso de precipitados y se agregó cloruro de calcio anhidro para secar la mezcla de reacción. Después se procedió a agregar carbonato de potasio para neutralizar la acidez remanente y se evaporó el solvente. Se obtuvieron 10 g de un sólido 9 en un rendimiento del 92 %. 9: Sólido p.f. 37-39 °C; RMN 1H (CDCl3, 200 MHz) δ: 3.75 (3H, s, p-CH3O-); 3.8 (6H, s, m-CH3O-); 4.5 (2H, s, Ar-CH2-); 6.55 (2H, s, Ar-H); IR (KBr) νmax: 3032, 2952, 1608, 1254, 1179, 1108, 829, 792, 717, 598 cm−1.
j) 3,4,5-Trimetoxibencil, 3-Etil-3hidroximetiloxetanil éter (10). En un matraz de tres bocas de 50 mL, provisto de condensador, termómetro y agitación magnética, se agregaron 3 g (0.0138 mol) del compuesto 9, 1.78 g (0.0152 mol) de 3 Etil-3-hidroximetiloxetano, y 0.66 g (0.0166 mol) de hidróxido de sodio, en 30 mL de dimetil sulfóxido. La reacción se mantuvo a 50 °C por espacio de 24 h. Después de finalizar este tiempo, se vertió la mezcla en 200 mL de agua destilada y se agregaron 100 mL de diclorometano. Enseguida se separaron las fases y la orgánica se lavó con agua destilada hasta eliminar completamente el DMSO. La fase orgánica se secó con sulfato de sodio anhidro y se evaporó para obtener un líquido incoloro que se purificó por cromatografía en columna usando cloruro de metileno como eluente. Se obtuvieron 3.2 g (78 %) del producto purificado. 10: Líquido incoloro p.eb. 198-205 °C a 0.1 mmHg; RMN 1H (CDCl3, 200 MHz) δ: 0.80 (3H, t, CH3-); 1.70 (2H, c, -CH2-); 3.50 (2H, s, -CH2-O-); 3.75 (3H, s, p-CH3O-), 3.8 (6H, s, m-CH3O-); 4.39 (4H, dd, -CH2O-CH2-); 4.45 (2H, s, ArCH2); 6.5 (2H, s, Ar-H); IR (KBr) νmax: 3054, 2936, 1641, 1243, 1182, 1125, 1050, 1012, 985, 926, 829, 786, 588, 528 cm−1 Anál. C 64.2 %, H 8.4% calcd para C 64.8 %, H 8.2 %.
Discusión y Resultados
En la Tabla 1 se muestra la estructura y el nombre químico de los cinco monómeros preparados. La estrategia para la síntesis de estos compuestos fue la de introducir al monómero un grupo que pueda estabilizar radicales libres, como sería en este caso el grupo bencílico. La estabilización de los radicales generados en la estructura del monómero, se debe tanto al efecto inductivo del oxígeno del éter como a la resonancia con el anillo aromático.
El diseño de estos nuevos monómeros involucra deliberadamente reacciones sencillas como sustituciones nucleofílicas y de oxidación, con el fin de hacer el proceso atractivo para una aplicación industrial. En la mayoría de las reacciones se hace uso de reacciones de eterificación, en la que se hace reaccionar un alcohol y un compuesto con un grupo saliente como puede ser un halogenuro de alquilo o un grupo p-toluensulfonato.
Por ejemplo, en el caso de la síntesis del monómero 3 se hizo reaccionar primero el alcohol 1,2,3,6-tetrahidrobencílico con el cloruro de p-toluensulfonilo para obtener el tosilato correspondiente 1. En seguida, se preparó el intermediario 2 por reacción entre 1 y el alcohol piperonílico en presencia de base, en sulfóxido de dimetilo como solvente. Se comprobó que al tratar de destilar el compuesto 2, este se descompuso por la acción del calor, generando presión. Por este motivo, se procedió a purificarlo por medio de cromatografía en columna. Una vez obtenido, se procedió a epoxidar el doble enlace usando un oxidante suave como el Oxone (2KHSO5. KHSO4.K2SO4), en condiciones neutras para evitar la hidrólisis del grupo acetal presente en el compuesto. Este monómero contiene tres centros quirales por lo que por RMN tanto de protón como de 13C se observa la presencia de estereoisómeros originados al oxidar el doble enlace
En la síntesis del compuesto 5 se utilizó el bromuro de tetrabutil amonio como catalizador de transferencia de fase para la eterificación del alcohol piperonilico con bromuro de alilo. Una vez obtenido el éter alílico se procedió a isomerizar el doble enlace mediante el uso del catalizador dicloruro de Tris-(trifenilfosfina)Rutenio ([(Ph)3P]3RuCl2). La isomerización fue seguida por RMN y se observó una conversión cuantitativa al éter propenílico 6. Se obtuvo una mezcla con una proporción 55:45 de isómeros cis:trans de acuerdo a su integración por RMN de protón. La asignación para cada protón fue determinada por RMN usando experimentos de doble irradiación sobre una determinada señal.
Dado que en este estudio se trató de preparar los principales tipos de grupos funcionales polimerizables catiónicamente, se decidió incluir monómeros de tipo oxetano. Esta funcionalidad oxetano es tan reactiva como los grupos epoxido al ser sometida a condiciones de fotopolimerización catiónica. La síntesis de estos monómeros fue reportada, por Crivello y Sasaki [16] e involucra la reacción entre el trimetilol propano y dietil carbonato, en presencia de carbonato de potasio como catalizador.
Esta reacción consiste de un intercambio de ésteres entre el trimetilol propano y el dietil carbonato para dar un carbonato cíclico de seis miembros, eliminando etanol al mismo tiempo que se forma. Este intermediario cíclico no se aisló y al continuar el calentamiento éste sufre una reacción de eliminación de dióxido de carbono para generar el oxetano deseado 8 (Esquema 4).
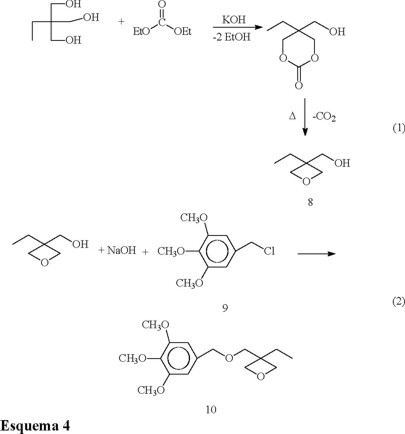
Este compuesto (3-etil-3-hidroximetil oxetano) se hizo reaccionar con el cloruro de 3,4,5-trimetoxibenzilo (9) e hidróxido de sodio en dimetil sulfóxido para dar el compuesto 10.
Conclusiones
Se reporta la síntesis de cinco nuevos monómeros que pueden polimerizar catiónicamente al ser irradiados por luz UV. La característica principal de estos diferentes tipos de monómeros entre los que se cuentan epoxiciclohexanos, vinil éteres, propenil éteres y oxetanos, es que contienen un grupo bencílico con grupos alcóxido. La activación del anillo aromático con estos grupos alcóxido, promueve una mejor estabilización del radical que se forma en la posición bencílica del monómero al ser extraído un átomo de hidrógeno por los radicales arilo formados durante la fotólisis del fotoiniciador. Posteriormente, éste radical bencílico es oxidado rápidamente por la misma sal de diariliodonio usada como fotoiniciador, para formar un carbocatión bencílico, que puede iniciar una polimerización catiónica. Este ciclo se repite en una reacción en cadena generando un gran número de especies catiónicas, lo que trae consigo un incremento en la velocidad de polimerización.
Las reacciones químicas involucradas en la preparación de estos monómeros, son reacciones químicas sencillas como reacciones de eterificación y de oxidación, las cuales son potencialmente atractivas parea una aplicación industrial.
Referencias
1. Dietliker, K. Chemistry and Technology of UV & EB formulation for coatings, inks & paints, ed P.K.T. Oldring, SITA Technology Ltd, London 1991. [ Links ]
2. Crivello, J.V. Radiation Curing in Polymer Science and Technology, vol.2, ed. J.P. Fouassier and J.F. Rabek, Elsevier Science Publishers, London, 1993. [ Links ]
3. Crivello, J.V. UV Curing Science and Technology; ed. Pappas, S.P.; Technology Marketing Corp. Stanford, CT, 1978. [ Links ]
4. Crivello, J.V. En Developments in Polymer Photochemistry-2; ed. N.S. Allen; Applied Sciences: London 1981, cap. 1. [ Links ]
5. Smith, G.H. US Pat. 4 394 403, July 19, 1983, Belgium Pat. 828 841, Nov. 7 1975. [ Links ]
6. Crivello, J.V. US Pat 3 981 897, Sept. 21, 1976 [ Links ]
7. Crivello, J.V.; Acosta, R.; J. Polym. Sci. Part A: Polymer Chemistry, 2001, 39, 2385-2395. [ Links ]
8. Ledwith, A. Polymer 1978, 19, 1217-1219. [ Links ]
9. Abdoul-Rasoul, F.A.M.; Ledwith, A.; Yagci, Y. Polymer 1978, 19, 1219-1225. [ Links ]
10. Bi, Y.; Neckers, D.C. Macromolecules 1994, 27, 3683-3693. [ Links ]
11. Crivello, J.V.; Jo, K.D. J. Polym. Sci., Polym. Chem. Ed. 1993, 31, 2143-2152. [ Links ]
12. Rajamaran, S.; Mowers, W.; Crivello, J.V. Macromolecules 1999, 32, 36-47. [ Links ]
13. Crivello, J.V.; Liu, S. J. Polym. Sci., Part A: Polymer Chemistry, 2000, 38, 389-401. [ Links ]
14. Crivello, J.V.; Liu, S. Chem. Mat. 1998, 10, 3724-3731. [ Links ]
15. Crivello, J.V.; Acosta, R. J. Polym. Sci. Part A: Polymer Chemistry, 2001, 39, 3578-3592. [ Links ]
16. Crivello, J.V.; Sasaki, H. J. Macromol. Sci. Pure Appl Chem. 1993, A30 (2&3) 173-187. [ Links ]

