



![Photochemistry of 7-Alcoxy and Thioalcoxy-3,3-dimethoxybicyclo[2.2.2] oct-5-en-2-one. Sequence 1,3-acyl shift-decarbonylation reaction](/img/es/prev.gif)






Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 no.3 Ciudad de México jul./sep. 2003
Investigación
La cinética de reacción y su influencia en la distribución de pesos moleculares, en un sistema de polimerización en emulsión sembrado
José Antonio Arcos Casarrubias,1* Roberto Olayo,2 Jaime Noriega Bernechea3
1 Subdirección de Investigación, Tecnológico de Estudios Superiores de Ecatepec, Av. del Mayo esq. Av. Hank González, Col. Valle de Anáhuac, Ecatepec, Edo. de México, CP 55210. Tel (55) 5710 4560. E-mail: jaac@tese.edu.mx
2 Departamento de Física, Universidad Autónoma Metropolitana-Iztapalapa, Ap. Postal 55-534, CP 09340; Tel/Fax: (55) 5804-4626. E-mail: oagr@xanum.uam.mx
3 Departamento de Fisicoquímica, Facultad de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, Coyoacán 04510, México. Tel/Fax: (55) 5622-3777.
Recibido el 12 de marzo del 2003
Aceptado el 22 de septiembre del 2003
Resumen
A partir de un modelo determinístico de la cinética de polimerización en emulsión, durante la etapa II de reacción, se muestra que la distribución de longitudes de cadena del polímero, P(N), es más sensible que la distribución de pesos moleculares W(M) para obtener información acerca del mecanismo que cesa el crecimiento de la cadena. La gráfica semilogarítmica de P(N) contra el grado de polimerización N, permite la evaluación de la relación entre los coeficientes cinéticos involucrados en la determinación de W(M). Las soluciones analíticas aproximadas de los sistemas de ecuaciones diferenciales y en diferencias resultantes, conseguidas mediante transformada Z, permiten el tratamiento de las tres etapas de polimerización en emulsión.
Palabras clave: Polimerización en emulsión, distribución de pesos moleculares, cinética de reacción.
Abstract
Based on a deterministic model of emulsion polymerization kinetics during stage II, we have shown that the polymer chain length distribution P(N), is more sensible than the molecular weight distribution W(M) to obtain information about the mechanism that stops chain growth. The semilogarithmic plot of P(N) against N, the extent of polymerization, permits the evaluation of the relationship among the kinetic coefficients involved in the determination of W(M). Approximate analytical solutions of the differences and differential equations systems were obtained by the Z transform; this approach can be applicable to all three emulsion polymerization stages.
Keywords: Emulsion polymerization, molecular weight distribution, reaction kinetics.
Introducción
Actualmente hay un interés renovado por desarrollar métodos precisos para la determinación de las constantes cinéticas de los sistemas de polimerización vía radicales libres, en particular, las constantes de terminación bimolecular y de transferencia de cadena. Una de las justificaciones de esto, es el interés cada vez mayor, de producir polímeros con distribución de pesos moleculares controlada. Así, el desarrollo de materiales con propiedades deseables, como son la distribución de pesos moleculares, la polidispersidad, la composición, los grupos funcionales y la arquitectura de la molécula con morfologías de orden de los nanómetros están en plena consolidación. Lo anterior está siendo posible debido a un método novedoso de síntesis de homopolímeros, y particularmente de copolímeros, llamado polimerización radicálica controlada o viviente [1, 2].
Con el propósito de obtener un producto con propiedades controladas, es imprescindible el manejo de los procesos que cesan el crecimiento de la cadena polimérica, a partir de principios científicos y no sólo del conocimiento empírico. En este sentido, actualmente se está trabajando en la dirección de esclarecer las cinéticas de polimerización, particularmente orientados hacia la reacción de terminación [3]. Dado que la reacción de terminación no puede ser totalmente suprimida, sí es posible disminuir su intervención hasta el punto que su efecto sobre las propiedades del polímero sean marginales. Por lo tanto, es ineludible el entendimiento básico de los mecanismos que ocasionan la cesantía del crecimiento de la cadena, principalmente los relativos a la terminación bimolecular [4-9]. En relación con la constante de transferencia, hay una necesidad de producción de polímeros de bajo peso molecular cuya aplicación está destinada a los recubrimientos (coatings) con un alto contenido de sólidos; con este propósito, el uso de agentes de transferencia de cadena es imprescindible, y por tanto, se impone la determinación de las constantes de transferencia al agente particular [10-14].
No obstante la enorme importancia técnica de la polimerización vía radicales libres, existe una dispersión muy grande de los valores tabulados de las constantes de rapidez, aún para sistemas de polimerización cuyas condiciones de reacción son ostensiblemente idénticas. Compilaciones exhaustivas de las diferentes constantes cinéticas, publicadas en la literatura, no han sido objeto de una evaluación crítica y adolecen de inconsistencias. Esta aseveración tiene sustento en la inspección efectuada a la tercera edición del Manual de los Polímeros de Brandup e Immergut [15].
Asimismo, existen en la literatura numerosos modelos de las cinéticas de polimerización vía radicales libres y de su influencia sobre la distribución de pesos moleculares [4,16-21]. Sin embargo, éstos modelos no han sido exitosos en la evaluación, particularmente de la constante de terminación, debido a que tienen un gran número de parámetros libres, y por tanto, no hay certeza de cuál mecanismo es el dominante en la distribución de pesos moleculares, y por lo general, sólo son un buen ejercicio de ajuste de datos a los modelos. En consecuencia, estos valores, cuando se han usado en modelos diferentes, aún con hipótesis contradictorias, pueden reproducir bien el comportamiento de los sistemas de polimerización vía radicales libres. Por lo tanto, es necesario el desarrollo de un modelo para condiciones de reacción muy particulares, en donde se conozca con certeza el mayor número posible de los parámetros del mismo, con el propósito de evaluar alguno de ellos.
En este sentido se ha desarrollado la técnica de polimerización mediante pulsos de láser y su posterior análisis de la distribución de pesos moleculares [22-25] y la técnica de polimerización por un pulso de láser y el seguimiento en línea del avance de la reacción mediante espectroscopía de infrarojo [25-27]. Otro enfoque es el propuesto por Clay y Gilbert, [28] quienes desarrollaron un modelo mecanístico detallado de la distribución de pesos moleculares.
La etapa II de la polimerización en emulsión está caracterizada por la ausencia de nucleación, que experimentalmente puede conseguirse a través de depositar una semilla en el sistema previamente al inicio de la polimerización. Esta etapa es particularmente útil para la evaluación experimental de los coeficientes de los procesos de pseudo primer orden [29] como son: la captura de radicales libres (ρ), la desorción de los radicales de las partículas (κ) y la terminación (c); además, se puede evaluar la constante de propagación (kp). La IUPAC recomienda esta técnica para la determinación de la constante de transferencia de cadena (ktr) [30].
Las ecuaciones de Smith-Ewart [31] establecen el balance de población de partículas que contienen un número determinado de radicales libres y constituyen el modelo cinético de reacción. Gilbert y colaboradores [32-34] han dado soluciones analíticas aproximadas al sistema dinámico correspondiente a los casos cinéticos I (n < 0.5) y II (n = 0.5), donde n es el número promedio de radicales por partícula. En estas soluciones mostraron que los coeficientes cinéticos del modelo están relacionados con la pendiente y la ordenada al origen de la gráfica de conversión contra tiempo; sin embargo, la evaluación experimental de dichos coeficientes presenta inconvenientes. No obstante que la pendiente puede ser medida con precisión, la ordenada al origen está afectada por gran incertidumbre, debido principalmente a dos cuestiones: por un lado no se conoce con certeza el origen del periodo de acercamiento al estado estacionario, lo cual hace muy difícil determinar en qué momento éste ya se ha establecido. Por otra parte, la cercanía del intercepto al cero del origen del sistema coordenado de medición, hace que los errores aleatorios asociados al experimento tengan el mismo orden de magnitud que la variable buscada, generando poca confiabilidad en esta región experimental. Por lo anterior, se hace evidente la necesidad de disponer de otro procedimiento alterno para corroborar los valores de los coeficientes cinéticos estimados. En este sentido, la distribución de pesos moleculares del polímero (DPM) es útil dado que es un registro de los procesos cinéticos involucrados en la formación del polímero y, por tanto, contiene información acerca de los coeficientes que caracterizan a dichos procesos.
Con esta intención, Clay y Gilbert [28] propusieron un modelo de la distribución de pesos moleculares basado en el modelo cinético de la polimerización en emulsión de Casey y colaboradores [35]. El modelo matemático resultante de la distribución de cadenas de radicales libres (DCR) y de la DPM son dos conjuntos de ecuaciones diferenciales y en diferencias fuertemente acopladas. Para su solución se recurre a la técnica de aproximación de variable discreta a variable continua; además, se usa la hipótesis de estado estacionario de la DCR, con lo cual se facilita el manejo matemático del sistema. Este modelo matemático trasformado está constituido por un conjunto de dos ecuaciones integrodiferenciales acopladas de la DCR y de la DPM, los cuales no tienes solución analítica. Sin embargo, los autores postulan diversas hipótesis con el propósito de encontrar soluciones analíticas aproximadas a diferentes situaciones límites; así, muestran la manera de obtener información mecanística y la posibilidad de evaluar algunos coeficientes cinéticos a partir de la medición de la DPM. Sin embargo, con excepción del coeficiente de terminación que se considera dependiente de la longitud de cadena, los coeficientes cinéticos ρ, κ y c son constantes y no dependen del tamaño de partícula.
En este trabajo se obtiene un modelo de la cinética de reacción de la polimerización en emulsión de un sistema sembrado; las ideas básicas del esquema cinético que consideramos están contenidas en el trabajo de Casey y colaboradores [35]. El modelo consiste de dos sistemas de ecuaciones diferenciales y en diferencias, que están fuertemente acopladas y cuyo número es infinito. Para la obtención de nuestras soluciones analíticas aproximadas, se acopló el balance de cadenas de radicales libres a las soluciones de los casos cinéticos I y II de Smith-Ewart [31-34]. Mediante el uso de la transformada Z se logró obtener la funcionalidad con el tiempo, tanto de la conversión así como de las DCR y DPM.
Una ventaja de disponer de la solución del sistema transiente es que, si se consideran intervalos de tiempo cortos donde efectivamente ρ, κ y c son constantes, entonces se pueden obtener la conversión y las DCR y DPM por intervalos; de modo que cuando se suman estos intervalos cortos se está abarcando el intervalo completo de polimerización.
Modelo cinético de la reacción de polimerización
Los supuestos contenidos para el establecimiento del modelo cinético son:
1) Sistema sembrado con tamaño monodisperso de partícula, lo cual elimina la posibilidad de la nucleación secundaria.
2) Sistema "cero-uno": el ingreso de algún radical libres a una partícula que previamente contiene uno, redundará en que la partícula quede sin radical porque la terminación bimolecular ocurre inmediatamente; por lo tanto, para partículas con diámetro pequeño es muy poco probable que puedan existir dos o más radicales dentro de una misma partícula de polímero.
3) La polimerización ocurre homogéneamente en el interior de las partículas de polímero; esta situación sucede cuando el polímero y el monómero son miscibles, la viscosidad en el interior no cambia apreciablemente con la conversión y cuando el tamaño de partícula sea pequeño, es decir cuando los efectos difusivos no sean significativos. En este caso, la constante de propagación de la reacción kp debe ser equivalente a la correspondiente de los sistemas en masa a la misma conversión.
4) El proceso de captura de radicales libres por las partículas es de pseudo primer orden con respecto a la concentración de radicales libres en las partículas; se incluye a los radicales libres generados por iniciador químico y a los radicales desorbidos de las partículas: ρ = ρI + ρre.
5) La única especie capaz de desorberse de las partículas es el radical libre monomérico; dicho radical se origina en el proceso de transferencia de la actividad de una cadena en crecimiento hacia un monómero. El radical de este tipo con dos o más unidades de monómero, la posibilidad de desorción es prácticamente nula.
Los mecanismos de reacción considerados para la derivación del modelo son:
a) Iniciación: se debe tanto a la captura de radicales libres originados por iniciador químico como a los radicales monoméricos. Es el paso que inicia la polimerización.
b) Propagación de la reacción: se debe a la unión química entre los monómeros, lo que provoca el crecimiento de la molécula.
c) Terminación bimolecular: se presenta cuando la partícula captura algún radical libre, verificándose una reacción instantánea y, por tanto, la cesantía del crecimiento de la cadena.
d) Transferencia de cadena: El sitio reactivo del radical libre se transfiere a otra molécula, ocasionando la cesantía del crecimiento de la cadena de la molécula que lo contenía y provocando el establecimiento de un nuevo radical. El proceso de transferencia de cadena a monómero es la fuente de radicales monoméricos.
A continuación, se procederá a establecer las ecuaciones de variación de monómero, de longitudes de cadena de los radicales y polímero muerto.
Consumo de monómero en las partículas de látex
Durante la etapa II, la concentración de monómero en el interior de las partículas tiene una dependencia débil con la conversión [36] y, por lo tanto del tiempo. Lo anterior es consecuencia de la concentración de equilibrio del monómero en la fase acuosa [29]. En nuestro trabajo, suponemos constante la concentración de monómero en el interior de las partículas. Con base en los supuestos y mecanismos anteriores, el balance de monómero en el interior de las partículas es (el tratamiento más detallado puede encontrarse en la referencia 37).
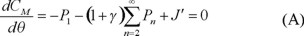
con  y
y 
CM es la concentración de monómero en el interior de las partículas, J es el flux de monómero hacia las partículas, Pn es la concentración de polímero activo con grado de polimerización n y, t el tiempo.
Variación de la concentración de las cadenas en crecimiento (polímero activo)
El balance de masa de polímeros activos en el interior de las partículas origina la ecuación de variación de las cadenas de polímero en crecimiento.
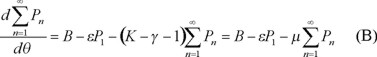
con 
y 
No es la concentración de núcleos sin radical, para cualquier tiempo.
Variación de la concentración de polímeros muertos
El balance de masa de polímeros muertos en las partículas da la ecuación de variación de las cadenas inactivas.
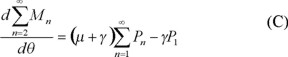
Mn es la concentración de polímero muerto con grado de polimerización n y, 
La concentración es proporcional al número de moléculas; por tanto, los sistemas de ecuaciones B y C son las tasas de variación de la población de cadenas de polímeros activos y las tasas de producción de la población de cadenas de polímeros muertos, respectivamente. La integración con respecto al tiempo de ambos sistemas de ecuaciones permite la obtención de las distribuciones de población acumuladas, tanto de polímeros en crecimiento como de los polímeros muertos.
La distribución de pesos moleculares está dada por la población de cadenas de polímeros muertos; para conocer la solución del sistema de ecuaciones C es necesario resolver previamente el sistema de ecuaciones B. Consecuentemente, se hace uso de la transformada Z [38, 39] para resolver el sistema de ecuaciones diferenciales ordinarias acopladas B.
La definición de dicha transformada es:

con lo cual, el sistema de ecuaciones B se reescribe
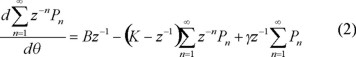
La ecuación ya transformada correspondiente a dicho balance es
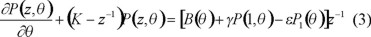
En principio, una solución numérica puede ser encontrada para esta ecuación. No obstante, se prefiere encontrar una solución analítica que permita conocer la forma funcional de la distribución de pesos moleculares como una consecuencia de los procesos de captura y desorción de radicales, propagación, transferencia y terminación bimolecular; con lo cual se podrá conseguir entender la influencia de los procesos químicos y físicos mencionados anteriormente. Para tal propósito se han considerado las siguientes situaciones límites: (1) Todos los radicales libres monoméricos se desorben y (2) ningún radical libre se desorbe. La primera situación corresponde al caso cinético I, mientras que la segunda al caso cinético II.
Caso cinético I
El proceso de salida de radicales libres de las partículas es muy rápido, de manera que una vez formado el radical libre monomérico, éste se desorbe inmediatamente de la partícula y no ocurre la propagación como tampoco la terminación bimolecular. La tasa de transferencia es igual a la rapidez del proceso de salida de radicales. Así, el paso lento es la formación de radicales libres monoméricos. Las hipótesis del caso cinético I son:
1) El valor del coeficiente de desorción de radicales es mucho mayor que el correspondiente al de la constante del proceso de terminación bimolecular: kex >> kt.
2) La tasa de captura de radicales libres es baja, lo cual implica que k > ρ >> c
3) Todos los radicales libres generados por transferencia de cadena se desorben de las partículas.
4) El paso lento es la formación de radicales libres monoméricos.
5) La tasa de transferencia de cadena es igual a la rapidez del proceso de salida. Es decir  y
y 
Por tanto 
Dividiendo entre kpCM se obtiene 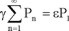
6) Es muy poco probable que puedan existir dos o más radicales libres por partícula.
7) La salida de radicales libres de las partículas es el fenómeno dominante.
Por lo tanto, la ecuación de balance de longitudes de cadena de polímeros activos se reduce a:
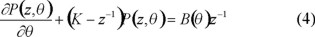
con la condición inicial P(z, θ) = 0 a θ = 0.
Nótese que B(θ) está dado por la solución de Gilbert y Napper [32] a las ecuaciones del balance de población de radicales libres en las partículas, para el caso cinético I de Smith y Ewart [31].

Con la finalidad de obtener una expresión más sencilla, se considera que B es independiente de θ, situación que se justifica por el hecho de que, en general, el establecimiento del estado estacionario es muy rápido comparado con el tiempo total de polimerización. Entonces, la ecuación a resolver es con la condición inicial P(z, θ) = 0 a θ = 0.
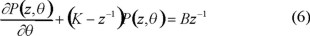
Desarrollo
La solución se encuentra por el método del factor integrante, obteniéndose
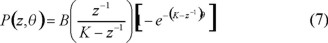
Ahora se requiere encontrar la transformada inversa, para lo cual la expresión anterior se desarrolla en series de potencias de z−1/K, puesto que |z−1/K| ≤ 1, de modo que se obtiene

mediante la comparación término a término entre la serie anterior y la definición de la transformada Z [38,39], se concluye que el número de cadenas de polímero activo con grado de polimerización n está dado por
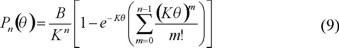
Dado que la transformada Z es, en el sentido estadístico, una función generadora de momentos, por consiguiente, la ecuación 8 es la función generadora de momentos de la distribución de cadenas radicales libres. Consecuentemente, su primer momento corresponde con la distribución acumulada de las cadenas de polímero en crecimiento.
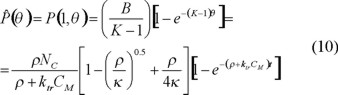
La ecuación anterior representa la totalidad de los radicales presentes en el sistema; para obtener su número promedio por partícula se divide entre el número de partículas. Por tanto, la conversión está dada por
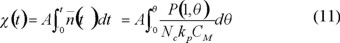

Finalmente, en función de los coeficientes cinéticos y del tiempo.
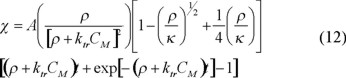
Ahora se puede resolver el conjunto de las ecuaciones que describe el balance de longitudes de cadena de polímeros muerto; nuevamente se hace uso de la transformada Z, de tal forma que dicho sistema se escribe como
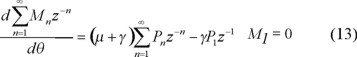
la cual se transforma a la siguiente ecuación

en donde se sustituyen las ecuaciones (7) y (8) con n = 1, obteniéndose

con la condición inicial M(z, θ) = 0 a θ =0. Se separan variables y se integra, obteniéndose
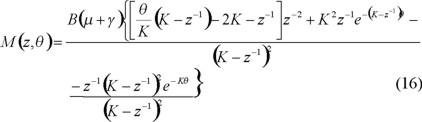
Nótese que la ecuación anterior (y también la 6) es, en el sentido estadístico, una función generadora de momentos; por tanto, los momentos y los valores promedios de la distribución se pueden obtener a partir de ésta. Así, el momento cero de la distribución, el cual representa la distribución acumulada de las cadenas de polímero muerto, es

y los momentos superiores se pueden obtener por diferenciación logarítmica.
Ahora se encuentra la transformada inversa [37], de tal modo que el número de cadenas de polímero con grado de polimerización n está dada por
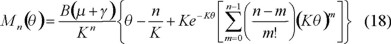
La distribución de longitudes de cadena (DLC) está relacionada de manera directamente proporcional con la distribución de pesos moleculares (DPM) a través de la masa molecular del monómero, M0. La distribución diferencial normalizada de la longitud de cadena (DLC) está dada por
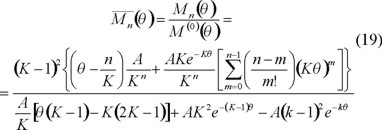
y representa a la fracción de moléculas con grado de polimerización n presentes en el sistema. Por otra parte, en la distribución diferencial normalizada de la longitud de cadena, el término

se puede ignorar porque exp(-kθ ) → 0 rápidamente. Se obtiene el logaritmo, lo que resulta en

Una gráfica de ln[Mn(θ)] contra n, resultaría ser lineal para tiempos grandes (altos pesos moleculares cuando θ >> n/K). En estas circunstancias, n es poco significativo en el término ln[A(θ − n/k)]. Por consiguiente, la pendiente únicamente está definida por −ln K, y aplicando la definición de K (dada en la ecuación (B)) se obtiene
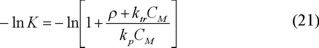
la expresión anterior se desarrolla en serie de potencias, obteniéndose

se ignoran los términos cuadrático y superiores, puesto que

Para este caso cinético I de Smith y Ewart [32,33], los valores de ρ son menores por tres órdenes de magnitud que los de ktr. Entonces, la pendiente está dada por
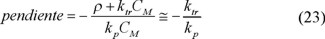
En este caso la transferencia de cadena es el único mecanismo que cesa el crecimiento de la cadena.
Caso cinético II
Para esta situación no hay desorción de radicales libres de las partículas y todos los radicales libres monoméricos inician el crecimiento de nuevas cadenas de polímero. El paso lento es el proceso de salida de radicales libres de las partículas. Las hipótesis del caso cinético II de Smith y Ewart [31] son:
1) La desorción de radicales de las partículas no ocurre: kex = 0.
2) La tasa de terminación bimolecular es mucho mayor que la tasa de captura de radicales: ρ << c.
3) Todos los radicales monoméricos generados por transferencia de cadena inician el crecimiento de nuevas cadenas de polímero.
4) El paso lento es la salida de radicales libres de las partículas: rex = − κ P1 con κ = kex / vs NA → 0.
Dividiendo entre kpCM se obtiene ε = κ / kp CM. Por tanto εP1 = 0.
5) El número promedio de radicales libres por partícula es
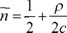
si ρ / 2c → 0, entonces nss = 0.5. Es el caso durante el estado estacionario
La ecuación de balance de masa de polímeros activos que describe el caso cinético II de Smith y Ewart es:
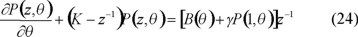
con la condición inicial P(z, θ) = 0 a θ = 0.
B(θ) está dada por la solución a las ecuaciones del balance de población de radicales libres en las partículas para el caso II de Smith y Ewart, elaborada por Hawkett y colaboradores [34]. Sin embargo, aquí se obtiene una relación relativamente sencilla para P(Z, θ) mediante la consideración que B no depende de θ; es decir que se considera su valor durante el estado estacionario. De esta manera la ecuación 21 se reduce a
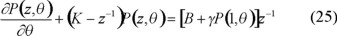
La dependencia de P(1, θ) con θ es  .
.
Mediante la sustitución de la expresión anterior en la ecuación (24) se obtiene
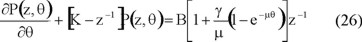
Nuevamente, aplicamos la metodología de la transformada Z al caso de la concentración de polímero activo con grado de polimerización n y se obtiene la expresión siguiente

y la concentración de polímero muerto con grado de polimerización n está dado por la expresión

La expresión anterior es una función generadora de momentos. Entonces, los momentos de la distribución se pueden obtener por el procedimiento ya establecido anteriormente.
La fracción mol de polímero muerto con grado de polimerización n es
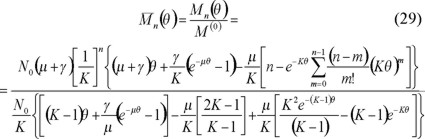
En la expresión anterior el término  se puede ignorar porque rápidamente θ → ∞ y entonces exp(−Kθ) → 0. Obteniendo logaritmos en ambos lados de la ecuación anterior
se puede ignorar porque rápidamente θ → ∞ y entonces exp(−Kθ) → 0. Obteniendo logaritmos en ambos lados de la ecuación anterior

Nuevamente se tiene la situación siguiente: una gráfica de contra n, resultaría ser lineal para tiempos grandes (región en la que predominan los altos pesos moleculares), cuya pendiente de la recta está dada por

mediante la expansión en serie de potencias del logaritmo, se ignoran los términos cuadrático y superiores, puesto que
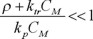
por lo que se llega a
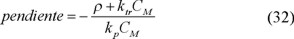
En el caso cinético II la distribución de pesos moleculares está controlada por la relación (ρ + ktrCM )/ kpCM, lo que sugiere que tanto la captura de radicales como la transferencia de cadena son procesos que cesan el crecimiento de las cadenas de los polímeros.
Resultados y discusión
La distribución de masas moleculares se obtiene a través de la relación

y que corresponde a la distribución medida por cromatografía de permeación en gel. Las Figs. 1A,B muestran la evolución de las distribuciones para los casos cinéticos I y II; en ellas se observa que la forma de la distribución se establece rápidamente, a tiempos cortos en un valor constante.

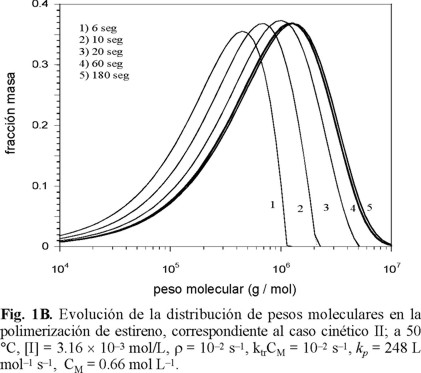
En la Fig. 2 se muestra la comparación entre las distribuciones de pesos moleculares para ambos casos cinéticos; en ella se aprecia que los pesos moleculares promedios son muy cercanos; sin embargo, la DPM correspondiente al caso cinético I presenta un sesgo hacia los pesos moleculares altos en referencia al caso cinético II, lo cual es indicativo de que el tiempo de vida promedio del radical libre dentro de la partícula, es mayor dado que la tasa de captura de radicales libres es baja y la desorción es significativa, con lo cual la tasa de terminación bimolecular es muy baja. El valor del índice de polidispersidad en ambos casos rápidamente tiende a 2 y prácticamente no varía; este es el valor esperado para la polimerización en emulsión durante la etapa II[29]. La figura 3 presenta la evolución del peso molecular numeral promedio para el caso cinético I, el cual se obtiene a través de la ecuación 14, y su comparación contra datos experimentales de Storti y colaboradores [40]; la concordancia entre el modelo y los datos experimentales es muy buena.
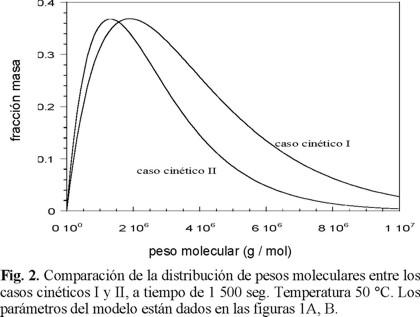

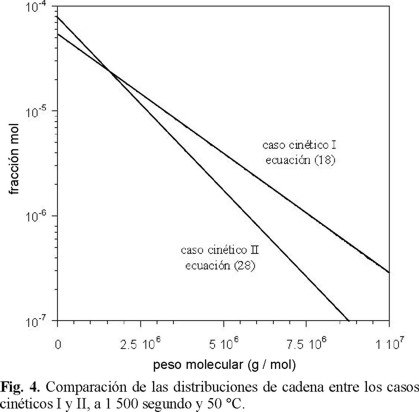
Las ecuaciones 18 y 28 representan la fracción mol de polímero que tiene grado de polimerización n, en función del tiempo y tiene como parámetros a los coeficientes ρ, ktr y kp, a partir de ellas se generan las distibuciones de longitudes de cadena correspondientes a los casos cinéticos I y II. La Fig. 4 presenta su comparación para el estireno a 50 °C, los valores de los parámetros cinéticos usados son: para el caso cinético I ρ es del orden de 10−4 s−1; y para el caso cinético II ρ = 10−2 s−1; así lo muestran los trabajos de Hawkett y colaboradores [36], Arcos [37] y Lansdowe [41]; el valor reportado por Tobolsky y Offenbach [42] de ktrCM es del orden de 10−2 s−1. El valor de kp ha sido establecido por un grupo de la IUPAC [43], aquí se utilizó 248 L mol−1 s−1; CM fue de 0.66 mol L−1. Como se esperaba, el valor de la pendiente para el caso cinético II es menor que el correspondiente al caso cinético I. Por consiguiente, la distribución de pesos moleculares está determinada por la relación ktr / kp, puesto que los valores de frente a los de ktrCM no son significativos; además, sugiere que el proceso de transferencia de cadena es el único mecanismo que cesa el crecimiento de la cadena de polímero. Por supuesto, los valores de ρ para el caso cinético II son mayores que los correspondientes al caso cinético I, y por lo tanto, los valores de las pendientes para el caso cinético II siempre son menores que los respectivos del caso cinético I. En el caso cinético II los valores de pueden ser del mismo orden de magnitud a los correspondientes de ktrCM. En esta situación la distribución de pesos moleculares está controlada por la relación, (ρ + ktr CM) / kp CM lo que sugiere que tanto la captura de radicales como la transferencia de cadena son procesos que cesan el crecimiento de las cadenas de polímero.
La consideración de un agente de transferencia de cadena modifica la expresión de la pendiente de la recta de la gráfica de ln[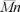 (θ)]contra n y está dada por
(θ)]contra n y está dada por
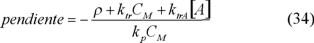
donde ktrA es la constante de transferencia al agente y [A] es la concentración del agente de transferencia dentro de la partícula.
La evaluación experimental de los coeficientes cinéticos mediante el análisis de la distribución de pesos moleculares está llevándose a cabo; no obstante, no existen muchas publicaciones en este sentido. Heuts et al. [44] comparan el método tradicional de Mayo [45, 46] contra el método de la distribución de pesos moleculares para la medición de constantes de transferencia, encontrando que ambos procedimientos son equivalentes y dan resultados confiables. Christie y Gilbert [47, 48] lo usaron para evaluar constantes de transferencia de metacrilato de metilo a diferentes agentes en la polimerización en masa.
Miller et al. [49] realizaron experimentos de polimerización en emulsión de estireno, en condiciones ab initio, a 70 °C. Estos autores encontraron que la distribución de pesos moleculares presenta, claramente, dos regiones: una lineal a altos pesos moleculares y otra cóncava a bajos pesos. Clay et al. [50] informaron de experimentos de polimerización en emulsión de estireno, sembrados con dos tamaños de látex; los cuales abarcaron hasta conversión de 80 %. Estos autores obtuvieron valores de <kt> en función de la conversión; sin embargo, dichos valores no concuerdan con los medidos por técnicas de relajación. Su explicación, para justificar esta diferencia, está sustentada en el flux de radicales libres producido en ambos tipos de experimentos, los cuales generan una población de radicales distinta.
Conclusiones
Mediante un modelo determinístico de la cinética de reacción en la etapa II de la polimerización en emulsión, se obtuvieron expresiones analíticas para la conversión, la distribución de masas moleculares de radicales y la distribución de masas moleculares de polímero en función del tiempo. Este tratamiento permitiría considerar la dependencia de los coeficientes cinéticos ρ, κ y c con el tamaño de partícula y con el avance de la reacción.
Se mostró que la distribución de longitudes de cadena P(N) del polímero formado es más sensible a los procesos cinéticos que la distribución de pesos moleculares W(M). Los experimentos consisten en la medición de concentraciones, las cuales son directamente proporcionales a la longitud de la cadena de polímero; en consecuencia, la forma funcional de la distribución de longitudes de cadena permite una evaluación más precisa de los parámetros cinéticos en el modelo de reacción y, por tanto, se puede obtener información acerca de los mecanismos que cesan el crecimiento de la cadena de polímero.
Las soluciones semianalíticas obtenidas sugieren condiciones experimentales bajo las cuales se debe efectuar un experimento de polimerización en emulsión, con el propósito de evaluar los diferentes coeficientes cinéticos. Se mostró que en el caso cinético I se puede estimar la relación de la constante de transferencia de cadena a la constante de propagación; en tanto que en el caso cinético II, además se puede estimar el cociente de la constante de terminación entre la constante de propagación.
Referencias
1. Matyjaszewski, K. En: Matyjaszewski K. editor, ACS symposium series 768, Washington, D. C.: American Chemical Society, 2000, 2-26. [ Links ]
2. Fukuda, T.; Goto, A. En: Matyjaszewski K. editor, ACS symposium series 768, Washington, D. C.: American Chemical Society, 2000. 27 - 38. [ Links ]
3. Buback, M. En: Matyjaszewski K. editor, ACS symposium series 768, Washington, D. C.: American Chemical Society, 2000, 39 -56. [ Links ]
4. Cárdenas, J. N.; O´Driscoll, K. F. J. Polym. Sci. Polym. Chem. Ed. 1976, 14, 883-897. [ Links ]
5. Adams, M. E.; Russell, G. T.; Casey, B. S.; Gilbert, R. G.; Napper, D. H.; Sangster, D. F. Macromolecules 1990, 23, 4624 -4634. [ Links ]
6. Russell, G. T.; Gilbert, R. G.; Napper, D. H. Macromolecules 1992, 25, 2459 - 2469. [ Links ]
7. Russell, G. T.; Gilbert, R. G.; Napper, D. H. Macromolecules 1993, 26, 3538 - 3552. [ Links ]
8. Scheren, P. A. G. M.; Russell, G. T.; Sangster, D. F.; Gilbert, R. G., German, A. L. Macromolecules, 1995, 28, 3637-3649. [ Links ]
9. Cunningham, M. F.; Mahabadi, H. K. Macromolecules 1996, 29, 835 - 841. [ Links ]
10. Davis, T. P; Kukulj, D.; Haddleton, D. M.; Maloney, D. R. Trends Polym. Sci. 1995, 3, 365-373. [ Links ]
11. Suddaby, K. G.; Maloney, D. R.; Haddleton, D. M. Macromolecules, 1997 30, 702-713. [ Links ]
12. Haddleton, D. M.; Maloney, D. R.; Suddaby, K. G.; Muir, A. V. G.; Richards, S. N. Macromol. Symp. 1996, 111, 37-49. [ Links ]
13. Heuts, J. P. A.; Kukulj, D.; Foster, D. J. ; Davis, T. P. Macromolecules 1998, 31, 2894-2905. [ Links ]
14. Kukulj, D.; Heuts, J. P. A.; Davis, T. P. Macromolecules 1998, 31, 6034-6014. [ Links ]
15. Brandup, J.; Immergut, E. H. Editores, Polymer Handbook, 3a. edición, Wiley Interscience, Nueva York, 1989. [ Links ]
16. Katz, S.; Shinar, R.; Saidel, G. M. Adv. Chem. Ser. 1969, 91, 145-157. [ Links ]
17. Min, K. W.; Ray, W. H. J. Macromol. Sci., Rev. Macromol. Chem. 1974, C11, 177-255. [ Links ]
18. Lichti, G.; Gilbert, R. G.; Napper, D. H. J. Polym. Sci., Polym. Chem. Ed. 1980, 18, 1297-1323. [ Links ]
19. Lichti, G.; Gilbert, R. G.; Napper, D. H. en Emulsion Polymerization; Piirma, I. editor; Academic: New York, 1982, p. 93. [ Links ]
20. Tefera, N.; Weickert, G.; Bloodworth, R.; Schweer, J. Macromol. Chem. Phys. 1994, 195, 3067-3085. [ Links ]
21. Ghielmi, A.; Storti, G.; Morbidelli, M.; Ray, W. H. Macromolecules. 1998, 31, 7172-7186. [ Links ]
22. Aleksandrov, A. P.; Genkin V. N.; Kitai, M. S.; Smirnova, I. M.; Sokolov, V. V. Kuantovaya Elektron, 1977, 4, 976-981. [ Links ]
23. Olaj, O. F.; Bitai, I.; Hinkelmann, F. Makromol. Chem., 1987, 188, 1689-1702. [ Links ]
24. Olaj, O. F.; Schnöll-Bitai, I. Eur. Poly. J. 1989, 25, 635-641. [ Links ]
25. Beuermann, S.; Buback, M. Prog. Polym. Sci. 2002, 27, 191-254. [ Links ]
26. Schweer, J. PhD Thesis, Göttingen, Alemania 1988. [ Links ]
27. Buback, M; Schweer, J. Z. Phys. Chem. N. F. 1989, 161, 153-165. [ Links ]
28. Clay, P. A.; Gilbert, R. G. Macromolecules 1995, 28, 552-569. [ Links ]
29. Gilbert, R. G. Emulsion Polymerization. A Mechanistic Approach; Academic Press, San Diego, 1987. [ Links ]
30. Buback, M.; Gilbert, R. G.; Russell, G. T.; Hill, D. J. T.; Moad, G.; O'Driscoll, K. F.; Shen, J.; Winnik, M. A. J. Polym. Sci., Polym. Chem. 1992, 30, 851-863. [ Links ]
31. Smith, W. V.; Ewart, R. W. J. Chem. Phys. 1948, 16, 592-599. [ Links ]
32. Gilbert, R. G.; Napper, D. H. J. S. C. Faraday I, 1974, 70, 391-399. [ Links ]
33. Hawkett, B. S.; Napper, D. H.; Gilbert, R. G. J. S. C. Faraday I, 1975, 71, 2288-2295. [ Links ]
34. Hawkett, B. S.; Napper, D. H.; Gilbert, R. G. J. S. C. Faraday I, 1977, 73, 690-698. [ Links ]
35. Casey, B. S.; Morrison, B. R.; Maxwell I. A.; Gilbert, R. G.; Napper, D. H. J. Polym. Sci., Polym. Chem. 1994, 32, 605- 630. [ Links ]
36. Hawkett, B.S.; Napper, D. H.; Gilbert, R. G. J.C.S. Faraday Trans. I, 1980, 76, 1323-1343. [ Links ]
37. Arcos-Casarrubias, J. A. Tesis de Maestría, Universidad Autónoma Metropolitana Iztapalapa, México, 1997. [ Links ]
38. Abraham, W. H. Ind. Eng. Chem. Fund., 1963, 2, 221-224. [ Links ]
39. Ames, W. F. Nonlinear Ordinary Differential Equations in Transport Processes; Academic Press, Londres, 1968. [ Links ]
40. Storti, G.; Polotti, G.; Carra S.; Morbidelli, M. Dechema-Monographien Band 127-VCH Verlagsgesellschaft, 1992, 407 -415. [ Links ]
41. Lansdowe, S.W.; Gilbert, R. G.; Napper, D. H.; Sangster, D. F. J.C.S. Faraday Trans. I, 1980, 76, 1344 - 1355. [ Links ]
42. Tobolsky, A.V.; Offenbach, J. J. Polym. Sci. 1955, 16, 311-314. [ Links ]
43. Buback, M.; Gilbert, R. G.; Hutchinson, R. A.; Klumperman, B.; Kuchta, F. D.; Manders, B. G.; O'Driscoll, ;K. F.; Russell G. T.; Schweer, J. Macromol. Chem. Phys. 1995, 196, 3267-3280. [ Links ]
44. Heuts, J. P. A.; Davis, T. P. ; Russell, G. T. Macromolecules 1999, 32, 6019-6030. [ Links ]
45. Odian, G. Principles of Polymerization, 2ª. Edición, Wiley, Nueva York, 1981. [ Links ]
46. Mayo. F. R. J. Am. Chem. Soc. 1943, 65, 2324 - 2329. [ Links ]
47. Christie, D. I.; Gilbert, R. G. Macromol. Chem. Phys. 1996, 197, 403-412. [ Links ]
48. Christie, D. I.; Gilbert, R. G. Macromol. Chem. Phys. 1997, 198, 663-663. [ Links ]
49. Miller; C. M.; Clay, P. A.; Gilbert, R. G.; El-Asser, M. S. J. Polym. Sci. A: Polym. Chem. 1997, 35, 989-1006. [ Links ]
50. Clay P. A.; Christie, D. I.; Gilbert, R. G., ACS Symposium Series 685, Washington, D. C.: American Chemical Society, 1998, 104-119. [ Links ]

