










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 no.2 Ciudad de México abr./jun. 2003
Investigación
Synthesis and Properties of 2-diazo-1-[2-(thiophen-2-ylmethoxy)-phenyl]-ethanone. Intramolecular Cyclization Through a Carbenoid Intermediate
Erick Cuevas Yañez,1 Abraham Arceo de la Peña,1 Joseph M. Muchowski2 y Raymundo Cruz Almanza1*
1 Instituto de Química, UNAM. Circuito Exterior, Ciudad Universitaria, Coyoacán, 04510, México, D.F. Tel. (52) (55) 5622-4408; Fax: (52) (55) 5616-2217. E-mail: raymundo@servidor.unam.mx
2 Roche Biosciences Department of Chemistry, Roche Palo Alto, 3431 Hillview Avenue, Palo Alto, CA 94304-1320, USA.
Recibido el 24 de junio del 2003.
Aceptado el 10 de julio del 2003.
Dedicated to Dr. Alfonso Romo de Vivar Romo.
Resumen
Se describe la síntesis de la 2-diazo-1-[2-(2-tiofenilmetoxi)-fenil]-etanona (1) la cual involucra la reacción de sustitución nucleofílica aromática del (2-tiofenil) metóxido de sodio con 2-flurorobenzaldehído, la oxidación en condiciones suaves del grupo aldehído al ácido carboxílico y posteriormente la transformación a α-diazocetona a través de un anhídrido carbónico-carboxílico como intermediario de reacción. El tratamiento del compuesto 1 con cantidades catalíticas de acetato de rodio (II) da la tiofenilmetil benzofuranona 10 por medio de una transposición sigmatrópica [2,3] del iluro de oxonio 9 derivado del carbenoide de rodio que se postula como intermediario de reacción.
Palabras clave: α-diazocetona, ciclización, carbenoide de rodio.
Abstract
The synthesis of 2-diazo-1-[2-(thiophen-2-ylmethoxy)-phenyl]-ethanone (1) is described. The procedure involves an aromatic nucleophilic substitution between the sodium 2-thiophenyl methoxide and 2-fluorobenzaldehyde, the subsequent mild-condition oxidation to the corresponding carboxylic acid, and final transformation through carbonic-carboxylic anhydride intermediate to the a-diazoketone. Treatment of compound 1 with catalytic amounts of rhodium (II) acetate gives the thienylmethyl benzofuranone 10 which comes from the [2,3] sigmatropic rearrangement of the oxonium ylide 9 derived from the rhodium carbenoid that is postulated as reaction intermediate key.
Key words: α-diazoketones, cyclization, rhodium carbenoid.
Introduction
The diazo group chemistry has taken a new interest as a consequence of the development of catalytic methods with transition metals that convert diazoketones in valuable tools in organic synthesis with several applications in homologations, X-H bond insertions, ylide formations, and reactions with alkenes and aromatic compounds [1].
Our interest has especially been focused in aryl and heteroaryl diazoketone synthesis since there are few examples that show the reactivity of this kind of compounds. Recently Doyle and coworkers [2] took advantage of the furan reactivity to prepare a macrocycle by the intramolecular insertion of the rhodium carbenoid from a w-furanyl diazoketone. On the other hand, Capretta and coworkers [3] reported that the treatment of diverse diazoalkanoyl thiophenes with catalytic rhodium (II) acetate gave the intramolecular cyclization compounds as major reaction products, and they isolated 5 and 6 mermbered-fused rings to thiophene.
Results and discussion
Taking the preceding reactions and as a part of a project that involves a study of the ω-diazoalkanoylthiophene reactivity, we were interested in synthesize a molecule which had in their structure a thiophene ring and a diazo group, in such a way they could react to each other to form 5 or 6 membered rings when a carbenoid from a rhodium or copper salt were generated. For this purpose we believed molecule 1 was appropriate for the study and we proceeded to prepare it.
Initially, we reacted the sodium salt of 2-hydroxymethyl-thiophene 2 with 2-fluorobenzaldehyde 3 to obtain the alkyl aryl ether 4 in 73 % yield through a nucleophilic aromatic substitution. Previously, the substitution on 2-fluorobenzaldehyde was reported as a process that readily occurs [4]. The 2-hydroxymethylthiophene used in this part was prepared by sodium borohydride reduction of thiophene-2-carboxaldehyde, which is commercially available [5].


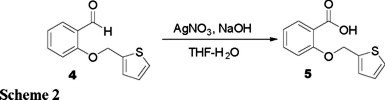

The next step in the synthetic route consisted on the aldehyde group oxidation in molecule 4 to the corresponding carboxylic acid. For this purpose, aldehyde 4 was treated with silver oxide, which was prepared in situ from the reaction of silver nitrate and sodium hydroxide. The oxidation reaction was performed at room temperature during 48 h in THF. After work up, benzoic acid 5 was obtained in 38 % yield.
Finally, the thiophen-2-ylmethoxy benzoic acid 5 became diazoketone 1 by the reaction with N-methylmorpholine and ethyl chloroformate in ether at 0 °C during 15 min, and subsequent addition of an excess of ethereal diazomethane (10:1) to the carbonic-carboxylic anhydride 6 which is postulated like the acylating agent. This technique has been successful in the synthesis of diverse pirrolyl diazoketones [6], and in this case gave quantitative yield.
Compounds 1, 2, 4 and 5 were characterized by the conventional spectroscopic techniques, and especially the diazoketone 1 was a crystalline solid which was studied by X-ray diffraction. The compound structure is represented in Fig. 2 and some important crystallographic data are in Table 1.

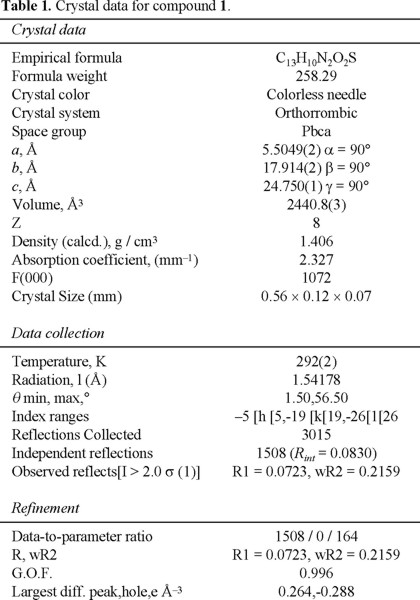
The X-ray study revealed some interesting data about the compound structure. For example, we observed that the angle formed between the nitrogen atoms of the diazo group is 177°, which indicates a strong tendency toward the lineal structure given by a resonant structure that favors two consecutive double bonds, one between C2 and N1 and the other between N1 and N2. Additionally, the bond distance between C1 and C2 is shorter than other distances with sp2 atoms, such as the C1-C3 bond distance. This can be explained by the resonance effects that should exist between the carbonyl group and the diazo group. In Table 2 some relevant angle and bond distances from the X-Ray study are presented for molecule 1.
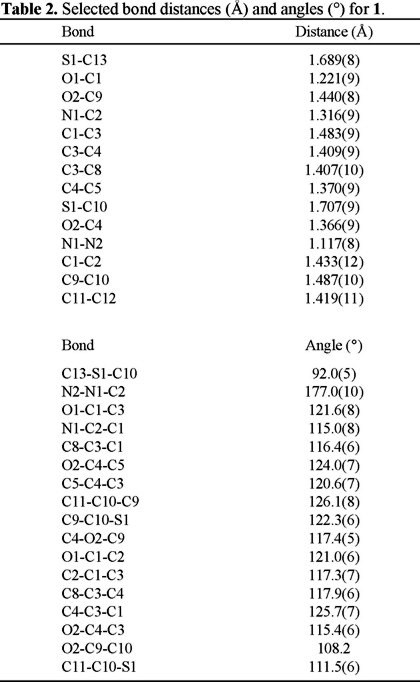
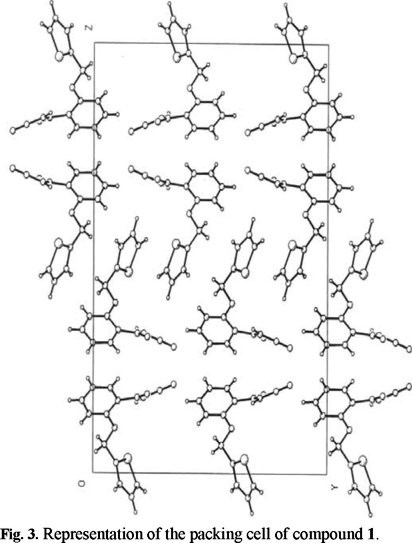
In the Fig. 3 we also could appreciate the packing cell representation for compound 1 that shows a great separation among the ring substituents. This suggests a strong repulsion between the ortho substituents which is probably due to the steric effect, and is reflected in the dihedral angle (C1-C3-C4-O2) with 8 ° instead of 0 °. Therefore, the substituents are not coplanar.
It is important to mention that there are not reported examples where a diazoketone has been studied and characterized by X-ray diffraction. Thus, the present work contributes with new data about the space distribution that presents this functional group.
Once prepared, diazoketone 1 reacted with catalytic amounts of rhodium (II) acetate. At the moment, when we carried out the reaction, we considered two possible reaction routes: the first consisted on the C-H insertion on the benzylic position of the carbenoid species 7 that it is formed from the diazoketone 1 and the catalyst, which would give thienylchromanone 8 as the major product; while in the second route we intended the formation of an oxonium ylide (9) after a sigmatropic rearrangement [2, 3] would afford benzofuranone 10. In this respect, we were not able to predict the reaction course because there are not overwhelming evidences about the reaction mechanism. The McKervey group [7-9] found that varying the catalyst they could modify the course of this kind of reactions to obtain both chromanones and benzofuranones. However, the five-membered ring formation and subsequent Stevens rearrangement is more common in these processes [10].
Treatment of diazoketone 1 with rhodium (II) acetate in dichloromethane at room temperature under inert atmosphere gave a reaction product whose physical and spectroscopic constants did not correspond to those expected for chromanone 8 as have been reported previously [11, 12]. In this way, spectroscopic data indicated that the compound obtained was thienylmetylbenzofuranone 10. Additionally, Gefflaut and Périe [13] informed a similar cyclization process when the diazoketone derived from 2-benzyloxybenzoic acid reacted with catalytic rhodium (II) acetate. Therefore, in this case the carbenoid 7 derived from the diazoketone 1 reacted in an intramolecular way with the phenoxy oxygen to generate a five-membered ring and also an oxonium ylide which is kinetically favored and is rearranged later to the benzofuranone 10.
In conclusion, this work presents the first synthesis of the 2-diazo-1-[2-(thiophen-2-ylmethoxy)-phenyl]-ethanone 1 and its intramolecular cyclization to the benzofuranone 10, which expands the possibilities to carry out reactivity studies with transition metals and to synthesize some derivatives from this molecule.
Experimental section
The starting materials were purchased from Aldrich Chemical Co. and were used without further purification. Solvents were distilled before use, ether and tetrahydrofuran (THF) were dried over sodium using benzophenone as indicator. Diazomethane was prepared from N-methyl-N-nitroso-p-toluenesulfonamide (Diazald ®) using a minimum amount of water and ethanol as co-solvent, and dried over KOH pellets before use. Silica gel (230-400 mesh) and neutral alumina were purchased from Merck. Silica plates of 0.20 mm thickness were used for thin layer chromatography. Melting points were determined with a Fisher-Johns melting point apparatus and they are uncorrected. 1H and 13C NMR spectra were recorded using a Varian Gemini 200, chemical shifts (d) are given in ppm relative to TMS as internal standard (0.00). For analytical purposes, mass spectra were recorded on a JEOL JMS-5X 10217 in the EI mode, 70 eV, 200 °C via direct inlet probe. Only molecular and parent ions (m/z) are reported. IR spectra were recorded on a Nicolet Magna 55-X FT instrument. For X-Ray diffraction studies, crystals of compound 1 were obtained by slow evaporation of a dilute ethanol solution, and reflections were acquired with a Nicolet P3 / F diffractometer. Three standard reflections every 97 reflections were used to monitor crystal stability. The structure was solved by direct methods, missing atoms were found by difference-Fourier synthesis, and refined on F2 by a full-matrix least-squares procedure using an isotropic displacement parameters using SHELX-97.
2-Hydroxymethylthiophene (2). To a suspension of sodium borohydride (0.339 g, 8.92 mmol) in absolute ethanol (9 mL) a solution of 2-thiophenecarboxaldehyde (2 g, 17.85 mmol) in ethanol (25 mL) was added maintaining the temperature below 25 °C. The resulting mixture was then heated at 50 °C during 1 h. The solvent was removed in vacuo, and water (50 mL) was added, the solution acidified with diluted HCl (10 %) to pH = 5. The product was extracted with ether (3 × 50 mL), the organic phase was dried over Na2SO4 and the solvent was removed in vacuo to yield a colorless oil (1.93 g, 95 %), which was used without additional purification. IR (CHCl3, cm−1). 3434, 2930, 1668. 1H NMR (CDCl3, 200 MHz) δ 2.25 (s, 1H), 4.79 (s, 1H), 6.98 (m, 2H), 7.33 (m, 1H). MS [EI+] m/z (%): 114 [M]+ (100), 113 [M - H]+ (40).
2-(Thiophen-2-ylmethoxy)benzaldehyde (4). To a suspension of sodium hydride (0.97 g, 20.3 mmol) in DMF (17 mL) a solution of 2-hydroxymethylthiophene 2 (1.93 g, 16.9 mmol) in DMF (17 mL) was added at 0 °C, the resulting mixture was stirred under a nitrogen atmosphere at room temperature for 15 min. The mixture was cooled at 0 °C and 2-fluorobenzaldehyde (2.2 g, 17.8 mmol) was added. The resulting mixture was stirred for additional 15 minutes at room temperature. The reaction was quenched by addition of water (100 mL) and diluted HCl (10 %) to pH = 5. The aqueous phase was extracted with ether (3 × 75 mL), the organic phase was washed with water (150 mL), dried over Na2SO4 and the solvent was removed in vacuo. Purification by column chromatography (SiO2, hexane / AcOEt 9:1) yield a colorless oil (2.67 g, 73 %). IR (CHCl3, cm−1). 3105, 1687. 1H NMR (CDCl3, 200 MHz) δ 5.35 (s, 2H), 7.07 (m, 3H), 7.35 (dd, 2H), 7.55(m, 1H), 7.85 (m, 1H), 10.5 (s, 1H). MS [EI+] m/z (%): 218 [M]+ (10), 97 [M-C7H5O2]+ (100).
2-(Thiophen-2-ylmethoxy)benzoic acid (5). Silver nitrate (8.4 g, 49.5 mmol) was added to a solution of sodium hydroxide (2.97 g, 74.2 mmol) in water (75 mL). The mixture was added to a solution of compound 4 (2.67 g, 12.3 mmol) in THF (14 mL) and the resulting mixture was stirred for 48 h at room temperature. The mixture was filtered and acidified to pH = 4, and the product was extracted with ethyl acetate (3 × 100 mL). The organic phase was dried over Na2SO4 and the solvent was removed in vacuo. The product was purified by crystallization (1.1 g, 38 %). m.p. 100 °C. IR (CHCl3, cm−1). 3511, 1664. 1H NMR (CDCl3, 200 MHz) δ 5.46 (s, 2H), 7.05 (m, 3H), 7.29 (dd, 1H), 7.45 (m, 1H), 7.6 (m, 1H), 8.22 (dd, 1H). MS [E I+] m/z (%): 234 [M]+ (7), 97 [M-C7H5O3]+ (100).
2-Diazo-1-[2-(thiophen-2-ylmethoxy)-phenyl]-ethanone (1). An ice-cold solution of the acid 5 (0.28 g, 1.2 mmol) in freshly distilled ether (2 mL) was treated successively with ethyl chloroformate (0.14g, 1.3 mmol) and N-methylmorpholine (0.12 g, 1.2 mmol), the mixture was stirred under nitrogen atmosphere for 15 min at 0 °C, then an ether solution of diazomethane (12 mmol) from N-methyl-N-nitroso-4-toluenesulfonamide was added at 0 °C. A vigorous evolution of nitrogen occurred, and the mixture was allowed to warm to room temperature overnight. The solvent was removed in vacuo and the product was purified by column chromatography (SiO2, hexane / AcOEt 9:1) to yield a yellow crystalline product (100 %).m. p 70 °C. IR (CHCl3, cm−1). 3139, 2104, 1762. 1H NMR (CDCl3, 200 MHz) δ 5.27 (s, 1H), 5.46 (s, 2H), 7.05 (m, 3H), 7.29 (dd, 1H), 7.45(m, 1H), 7.6 (m, 1H), 8.22 (dd, 1H). MS [EI+] m/z (%): 258 [M]+ (5), 120 [C7H4O2]+ (100).

2-(Thiophen-2-ylmethyl)benzofuran-3-one (10). A solution of the diazopropanone 1 (0.3096 g, 1.2 mmol) in dry CH2Cl2 (10 mL) was stirred with Rh2(OAc)4 (2 mg) under nitrogen atmosphere at room temperature. After 2 h, the mixture was evaporated in vacuo and purified by column chromatography (SiO2, hexane / AcOEt 9:1) to yield a white solid (0.118 g, 43 %). m.p. 45 °C. IR (CHCl3, cm−1). 2923, 1716, 1612. 1H NMR (CDCl3, 200 MHz) δ 3.30 (dd, 1H), 3.62 (dd, 1H), 4.78 (dd, 1H), 6.95-7.22 (m, 4H), 7.60-7.80 (m, 3H). MS [EI+] m/z (%): 230 [M]+ (15), 121 [C7H5O2]+ (100), 97 [C5H5S]+ (75).
Acknowledgments
Financial support from CONACyT (no. 27997E) is gratefully acknowledged. The authors would like to thank Rocío Patiño, Angeles Peña, Javier Pérez and Rubén A. Toscano for their technical support.
References
1. Doyle, M. P.; McKervey, M.A.; Ye, T.; Modern Synthetic Methods using Diazocompounds: from Cyclopropanes to Ylides, John Wiley & Sons: New York, 1998. [ Links ]
2. Doyle, M.P., Chapman, B.J.; Hu, W.; Peterson, C.S.; McKervey, M.A.; Garcia, C.F.; Org. Lett. 1999,1, 1327-1329. [ Links ]
3. a)Frampton, C.S.; Pole, D.L.; Yong, K.; Capretta, A. Tetrahedron Lett. 1997, 38, 5081-5084. [ Links ] b) Yong, K.; Salim, M.; Capretta, A. J. Org. Chem. 1998, 63, 9828-9833. [ Links ]
4. Yeager, G.W.; Schissel, D.N. Synthesis 1995, 28-30. [ Links ]
5. Gronowitz, S.; Liljefors, S. Chem. Scr., 1978-79, 13, 39-45. [ Links ]
6. Jefford, C.W.; Kubota, T.; Zaslona, A. Helv. Chim. Acta 1986, 69, 2048-2061. [ Links ]
7. Pierson, N.; Fernández-García, C.; McKervey, M. A.; Tetrahedron Lett. 1997, 38, 4705-4708. [ Links ]
8. Ye, T.; Fernandez-García, C.; McKervey, M. A.; J. Chem. Soc. Perkin Trans. 1, 1995, 1373-1379. [ Links ]
9. Ye, T.; McKervey, M.A. J. Chem. Soc. Chem. Commun. 1992, 823-824. [ Links ]
10. Hodgson, D. M.; Pierard, F. Y. T. M.; Stupple, P.A. Chem. Soc. Rev. 2001, 30, 50-61. [ Links ]
11. De, M.; Majundar, D. P.; Kundu, N. G. J. Indian Chem. Soc. 1999, 76, 665-674. [ Links ]
12. Corvaisier, A. Bull. Soc. Chim. Fr. 1962, 528-535. [ Links ]
13. Gefflaut, T.; Périe, J. Synth. Commun. 1994, 24, 29-33. [ Links ]

