










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.46 no.2 Ciudad de México abr./jun. 2002
Revisión
Análisis por inyección en flujo: herramienta clave para la automatización analítica
María del Pilar Cañizares Macías*
Facultad de Química, Universidad Nacional Autónoma de México. Ciudad Universitaria, Coyoacán 04510, México, D.F. Tel: 5622-3786; Fax: 5622-3723. E-mail: pilarm@servidor.unam.mx
Recibido el 30 de mayo del 2001.
Aceptado el 11 de enero del 2002.
Resumen
El análisis por inyección en flujo (FIA, por sus siglas en inglés: Flow Injection Analysis), en sus diferentes versiones, es una de las formas más avanzada que el químico analítico tiene a su disposición para manipular disoluciones acuosas o mezclar reactivos y así, sin intervención humana, finalmente transportar al lugar de medida los componentes de una o más reacciones químicas. En este trabajo se hace una revisión desde el inicio del Análisis por Inyección en Flujo hasta nuestros días y el futuro que le espera a esta técnica analítica que cada vez es más utilizada en los laboratorios de rutina de diversas áreas (clínica, farmacéutica, ambiental y de alimento), gracias a su gran versatilidad.
Palabras clave: Análisis por Inyección en Flujo, desarrollo del FIA.
Abstract
Flow Injection Analysis (FIA) in its different versions, is one of the most advanced technique that can be used to manage aqueous solutions or to mix reagents without the human hand by the analytical chemist and so, finally, carry to the measurement place the compounds of one or more chemical reactions. A revision from the beginning of the FIA until our days and the expected future for this analytical technique, which is more and more applied in clinical, pharmacological, environmental and food routine laboratories because of its good versatility, is made in this paper.
Keywords: Flow injection, development of FIA.
Introducción
El uso de los métodos automáticos de análisis va ligado al concepto de sustitución de una operación o medida estática realizada manualmente por una operación y medida continua o secuencial. Este concepto lo introdujo Skeggs en 1957 [1] al describir el primer sistema de flujo continuo segmentado empleando burbujas de aire para dividir el flujo de la muestra en muchas porciones individuales. Este sistema se utilizó, principalmente, en laboratorios de análisis clínicos en la década de los 60 cuando salieron al mercado los primeros autoanalizadores semiautomáticos. La importancia de estos métodos radicaba principalmente en la disminución de la manipulación del analista en el progreso de la reacción, obteniendo una metodología que solo podía ser "semiautomatizada" debido al poco desarrollo en los programas de computación capaces de procesar los datos obtenidos por el instrumento analítico, en muchos casos, los datos eran recolectados por medio exclusivamente de un graficador de carta.
Entre los métodos automáticos de análisis se encuentra el Análisis por Inyección en Flujo (FIA: flow injection analysis) concepto introducido por Ruzicka y Hansen en el primer artículo publicado en 1975 [2] aunque en 1970 Nagy, Feher y Pungor [3] describen el uso de electrodos de grafito para mediciones voltamperométricas de muestras inyectadas, por medio de una jeringa hipodérmica, dentro de un portador, sin segmentar el flujo. Este trabajo es quizás el primer informe acerca de lo que podría ser lo que posteriormente se llamo análisis por inyección en flujo. El impacto que esta técnica provocó en muchos investigadores resultó en el desarrollo de nuevas metodologías por FIA, especialmente en el análisis de aguas. Para 1979 se contaba ya con 80 publicaciones y hoy, a 25 años de los inicios del FIA, el número de publicaciones estimado es de 10,000.
Análisis por inyección en flujo
En la década de los sesenta el método automático continuo de análisis más utilizado fue sin duda el análisis por flujo segmentado (SFA: segmented flow analysis). En esta técnica las muestras son aspiradas secuencialmente y entre ellas se sitúan burbujas de aire que separan (segmentan) el flujo establecido, incluido un ciclo de lavado para evitar contaminación entre muestras; las burbujas de aire se eliminan antes de que alcancen la celda de flujo localizada en el detector. El análisis por inyección en flujo tiene su antecedente en el SFA; pero en FIA, a diferencia del SFA, el flujo no esta segmentado y la muestra es inyectada en lugar de aspirada, en el momento de la detección no se alcanza ni el equilibrio físico ni químico. En la Fig. 1 se muestran las configuraciones generales para FIA y SFA.

En los primeros trabajos de FIA se presuponía la existencia de un flujo turbulento para explicar la ausencia de contaminación mutua de dos muestras inyectadas sucesivamente en el flujo [2]. En trabajos publicados posteriormente White y sus colaboradores [4], Stewart [5] y Ruzicka y su grupo [6] demostraron que para sistemas de flujo continuo el número de Reynolds era casi siempre inferior a 2000, por lo que se puede asegurar que existe un régimen laminar en el transporte de materia a través de los tubos, mientras que para el SFA el número de Reynolds es mayor o igual a 3000, debido al régimen turbulento que se lleva a cabo.
La existencia de un régimen de flujo laminar proporciona importantes ventajas al FIA: a) zona de muestra bien definida, b) caudales reducidos, y por lo tanto, consumo pequeño de reactivos y c) alta frecuencia de muestreo, lo que simplifica el montaje al no ser necesario una presión elevada ni burbujas separadoras de aire para evitar la contaminación mutua de muestras al ser inyectadas sucesivamente.
Dispersión controlada: fundamento del FIA
Las condiciones de trabajo habituales del FIA ocasionan que el bolo de muestra inyectado no alcance el equilibrio químico ni físico, además de originar que este se diluya (disperse) en la disolución portadora o en el reactivo
El régimen laminar produce una zona bien definida del bolo de muestra inyectado produciéndose un gradiente de concentración a lo largo de todo el bolo. En la Fig. 2 se muestra la dispersión que sufre la muestra a lo largo de una configuración FIA, al ser un sistema continuo se obtiene una señal transiente cuyo máximo corresponde a la parte del bolo menos diluida.
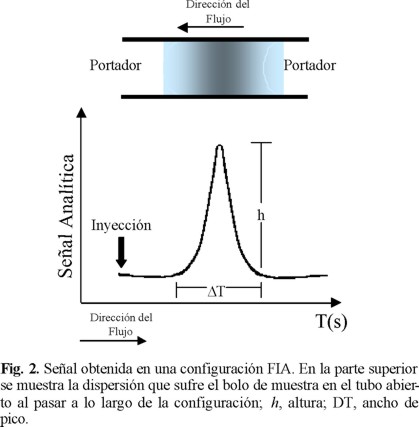
Existen dos mecanismos que contribuyen a la dispersión del bolo de muestra inyectado:
a) El transporte por convección desarrollado en condiciones de flujo laminar, que origina un perfil parabólico donde la velocidad lineal de las moléculas en las paredes del tubo es igual a cero, mientras que las moléculas que se encuentran en el centro del tubo viajan al doble de la velocidad media; y
b) El transporte por difusión debido a la formación de gradientes de concentración (horizontales y/o verticales) en diferentes momentos del transporte por convección.
Por lo tanto, la forma de una señal FIA (pico) dependerá del tiempo en el que el bolo de muestra se encuentra dentro del sistema. A tiempos demasiado cortos la dispersión es prácticamente cero y no se genera un gradiente de concentración en el bolo inyectado; por otra parte, tiempos de estancia largos ocasionan una dispersión muy acusada originando picos muy anchos. Debido a esto es necesario establecer cual es el tiempo de residencia más adecuado, siendo un parámetro difícil de decidir. Generalmente, la señal óptima es aquella en el que el transporte por convención y por difusión influye de la misma manera, no predominando uno sobre el otro, aunque evidentemente esta decisión dependerá de la reacción que se va a llevar a cabo.
Las características del pico (tiempos de arranque, residencia y ancho de banda y altura o área del pico) dependen de las características hidrodinámicas del sistema FIA, caudal, volumen de inyección, longitud del reactor, etc., siendo difícil establecer un modelo teórico de la dispersión capaz de predecir el comportamiento de un bolo inyectado en una configuración FIA. Generalmente los sistemas FIA tiene más de un canal por lo que debe tenerse presente las confluencias (tamaño y geometría), la geometría de la celda de flujo, las conexiones, el volumen de inyección, etc.
Diversos autores han discutido la aplicación de algunos modelos teóricos en las condiciones experimentales del FIA entre los que destacan el "modelo de Taylor" que solo es aplicable a caudales bajos y reactores de gran longitud [7, 8, 9, 10] y el "modelo de tanques en serie" utilizado por Ruzicka y Mottola que esta basado en el desarrollo de platos teóricos como en cromatografía líquida. La validez del modelo es dudosa cuando N (número de platos teóricos) es pequeño, lo que ocurre cuando la longitud del reactor no es extremadamente larga, lo que es habitual en FIA.
Es así como el coeficiente de dispersión de Ruzicka es el parámetro más adecuado para caracterizar la dispersión en un sistema FIA y fue propuesto por Ruzicka y sus colaboradores en 1975 [2]. Se trata de una medida directa de la dilución que sufre el trozo de muestra inyectado cuando pasa por el detector. Se define como la relación matemática de concentraciones de la muestra antes (Co) y después (C) de que haya tenido lugar el proceso de transporte en una configuración de FIA, es decir el inverso del factor de dilución:
D = Co / C
Este parámetro define el grado de dilución que ha sufrido una muestra en diferentes momentos del transporte. A cada punto del pico FIA le corresponde un coeficiente de dispersión, así en el máximo de la curva D = Co / Cmax.
Como siempre existe una relación entre la señal analítica detectada (absorbancia, milivoltios, intensidad de fluorescencia, etc.), el registro de la misma (altura) y la concentración de la muestra o de su producto de reacción (C) el coeficiente de dispersión puede calcularse a partir de la altura de la señal sin sufrir dispersión y de la señal registrada en el diagrama; de esta manera D = Koho / K1 hmax, donde Ko y K1 son las constantes de proporcionalidad que ordinariamente tienen el mismo valor ya que a las concentraciones trabajadas no varía la relación entre la concentración y la señal o altura. Por lo tanto el coeficiente de dispersión es una relación de alturas: D = ho / hmax.
Desde el punto de vista práctico la dispersión del bolo de muestra inyectado dependerá de las condiciones hidrodinámicas del sistema. La dispersión global del sistema puede considerarse como la sumatoria de las dispersiones originadas en las tres parte del sistema FIA: la dispersión originada tanto por el volumen de muestra como por los aspectos geométricos del sistema de inyección, la dispersión por la geometría del reactor y el caudal establecido y por la dilución provocada por la geometría de la celda de flujo. Con este principio se puede establecer, de manera general, que la altura de la señal FIA depende principalmente del tiempo de residencia del bolo de muestra inyectado en el sistema. A mayor tiempo de residencia mayor dispersión, por lo que reactores largos o caudales bajos incrementan la dispersión. Por otra parte, volúmenes de inyección pequeños incrementan la dilución de la muestra disminuyendo la altura del pico FIA.
Cuando ocurre una reacción química, considerar la dispersión como un fenómeno puramente físico como consecuencia de proceso de convección o difusión no es correcto. Los aspectos cinéticos deben ser considerados en FIA ya que la medición se realiza en condiciones de no-equilibrio, esta contribución no es tan significativa en SFA debido a que en este caso ya se ha alcanzado el equilibrio en el momento de la medición.
Una reacción química puede alterar el valor del coeficiente de dispersión en FIA de dos maneras: a) cuando se mide la propiedad del producto de la reacción que se produce en el flujo, que es la situación habitual; en este caso la contribución química hace que el valor de la dispersión práctica disminuya por lo tanto cuanto mayor sea la velocidad de reacción menor es la dispersión. b) Cuando se mide la propiedad de uno de los componentes de la reacción que desaparece en el flujo con el tiempo. En este caso la contribución química hace aumentar el valor del coeficiente de dispersión. Cuanto mayor sea la velocidad de reacción mayor es la dispersión. Painton y Mottola [11] han considerado este último caso en un artículo muy interesante donde determinan la contribución cinética, debido a la desaparición del dicromato, de la reacción entre el dicromato (muestra monitoreada a 352 nm) y el ácido ascórbico (portador) que está fuertemente influenciada por el pH ya que al disminuir el pH se aumenta la velocidad de reacción aumentando la dispersión.
Pasado, presente y futuro del análisis por inyección en flujo
Para 1975 Ruzicka y Hansen publicaron el primer artículo de una serie de informes sobre análisis por inyección en flujo. En este artículo describen los conceptos de los sistemas de flujo continuo segmentado y no-segmentado y sus diferencias. Para ejemplificar esas diferencias realizan la determinación colorimétrica de naranja de metilo, de fosfatos, por el método del azul de molibdeno, y la determinación electroquímica de amonio. En todas las determinaciones la frecuencia de muestreo era superior a las 200 inyecciones por hora con una precisión entre el 1 y 2 %. En el segundo artículo de esta serie Ruzicka y Stewart [12] describen los principales parámetros que se deben optimizar en un sistema FIA: volumen de muestra, diámetro del tubo, longitud del tubo, etc., ejemplificada por la determinación de fósforo en plantas.
Para 1979 Hansen y Ruzicka publicaron un artículo donde demostraban la confiabilidad del FIA para determinaciones analíticas para tres parámetros diferentes [13]: cloruros, fosfatos y una titulación de un ácido fuerte con una base fuerte. La determinación de cloruros se basa en la reacción con tiocianato de mercurio liberando tiocinato que reacciona cuantitativamente con Fe(III) formando un complejo color rojo que es proporcional a la concentración de cloruros en disolución. La configuración es muy sencilla, de un solo canal, obteniendo una frecuencia de muestreo de aproximadamente 120 / h con una precisión menor al 2 %. Para la titulación de un ácido fuerte con una base fuerte, las muestras ácidas se inyectan dentro del sistema de flujo continuo utilizando una cámara de mezclado para controlar el gradiente de dilución de las muestras. El portador utilizado es NaOH que contiene azul de bromotimol como indicador. En este caso no se mide la altura del pico FIA sino el ancho a una altura fija, generalmente, cuando se realizan titulaciones, el ancho del pico se mide a la mitad de éste.
En otras aplicaciones Krug y su grupo [14], realizaron la determinación turbidimétrica de sulfato en muestras acuosas en un intervalo de concentraciones de 40 a 140 ppm y con una velocidad de muestreo de 180 / h y precisión de 0.85 %. En este trabajo se presenta los efectos de la concentración del reactivo, el caudal del reactivo y la longitud de los reactores.
Como se puede ver los primeros trabajos realizados por FIA estuvieron enfocados a contaminantes en agua, debido a que se trata de una matriz fácil de trabajar y en muchos casos con pocas interferencias ya que generalmente no se trabajaba con aguas residuales. En la Tabla 1 se muestran algunas de las determinaciones que se han realizado por FIA en distintos tipos de agua.
Para mediados de la década de los 80' s las metodologías FIA ampliaron su campo de trabajo realizando determinaciones simultáneas y preconcentraciones en configuraciones cada vez mas complicadas pero más eficientes y con una alta probabilidad de automatización. Luque de Castro y su grupo [28] describen una configuración en la que un bolo de muestra queda dentro de un sistema cerrado permitiendo que éste pase repetidamente a través de un detector simple (espectrofotómetro) hasta que la muestra se dispersa casi en su totalidad dentro del portador. La señal obtenida esta dada por una serie de picos cuyos máximos o mínimos define la curva cinética de la reacción que se está llevando a cabo. Por medio de esta configuración es posible determinar los parámetros cinéticos de una reacción así como también la posible aplicación en métodos de concentración o dilución de muestras.
Este mismo grupo de trabajo [29] desarrolló, en 1987, un método para realizar la determinación simultánea de pH, alcalinidad y concentración iónica total en agua potable. El pH se midió por medio de un electrodo combinado de pH insertado en el flujo portador. La alcalinidad y los iones totales los determinaron por medio de dos titulaciones utilizando naranja de metilo para la determinación de alcalinidad y fenolfataleina para los iones totales, además para esta última determinación utilizan una microcolumna catiónica.
El uso de resinas y fases estacionarias en configuraciones FIA ha permitido que los niveles de concentración de los analitos determinados sean cada vez más pequeños ampliando el campo de automatización por medio del FIA. Este tipo de microcolumnas generalmente se han insertado en sistemas FIA acoplados a detectores de absorción atómica donde han tenido un gran desarrollo. Sven Olsen y sus colaboradores [30] utilizaron una microcolumna de Chelex 100 para pre-concentrar en línea plomo, cadmio y zinc, en muestras de agua de mar, alcanzando concentraciones de 10 ppb para plomo y de 1 ppb para los otros dos. La velocidad de muestreo fue entre 30 y 60 muestras por hora dependiendo del tiempo de pre-concentración requerido. En otro trabajo, Zhaolun Fang y su grupo [31] realizan el estudio de 122 resinas catiónicas débiles además de Chelex 100 y 8-quinolol para la pre-concetración de Cd, Zn, Pb y Cu, alcanzado niveles de hasta 0.03 µg / L para el Zn en 8-Quinolol.
La incorporación de microcolumnas en los sistemas FIA llevó consigo la incorporación de reactores enzimáticos aumentando la selectividad en muchas de las determinaciones que se habían estado realizando. La mayoría de las investigaciones están enfocadas a la inmovilización de la enzima en esferas de vidrio de poro controlado utilizada principalmente por su simplicidad y las buenas propiedades que ofrece el vidrio como matriz inerte, aunque los grandes avances que ha tenido la electroquímica ha permitido el desarrollo de sensores enzimáticos con mayor sensibilidad.
Uno de los primeros trabajos en considerar las enzimas inmovilizadas en configuraciones de flujo no segmentado fue el realizado por Gorton y Ogren [32] donde inmovilizan glucosa oxidasa y ureasa para la determinación de glucosa y urea en sangre y orina. Para la determinación de ambos parámetros en este tipo de muestras es necesaria una membrana dializadora situada entre el sistema de inyección y el reactor, con objeto de eliminar interferencias de matriz.
Diversos métodos potenciométricos para la determinación de parámetros clínicos han sido puestos a punto basándose en el uso de electrodos enzimáticos. Uno de los requisitos para que un sensor enzimático pueda utilizarse en un sistema de flujo es que la membrana sea resistente, ya que, además de soportar las características de la corriente, su manipulación es constante. Muchos métodos de inmovilización de enzimas para la construcción de electrodos proporcionaban membranas frágiles y la integridad mecánica de la membrana portadora de la enzima se dañaba después de un periodo corto de uso. Mascini y Palleschi [33] realizaron la determinación potenciométrica simultánea de glucosa y urea en suero inmovilizando las ånzimas sobre mallas de nylon que se fijan en sensores de oxígeno y amonio obteniéndose un buen funcionamiento y resistencia mecánica
Muchas de las investigaciones a lo largo de la década de los 80's estuvieron enfocadas a determinaciones enzimáticas por FIA principalmente con aplicaciones clínicas alcanzando un gran número de publicaciones. En la Tabla 2 se muestran algunos de estos ejemplos.
Ya para la última década del siglo XX el desarrollo en FIA estuvo enfocado principalmente al desarrollo de sensores químicos y bioquímicos (que incluye a los sensores enzimáticos) el acoplamiento de extracciones en línea (líquido-líquido, líquido-sólido, etc.) y el pre-tratamiento de muestras cn línea.
Los sensores en flujo donde se integra la separación con la detección se basan en el uso de resinas de intercambio o fases estacionarias como C18 que se empacan en una celda de flujo localizada en un detector no destructivo (por ejemplo fotómetro o fluorímetro) donde los analitos o los prjductos de reacción son inmovilizados temporalmente. Este tipo de sensores implica un gran intervalo de aplicaciones. Por ejemplo, el Cr(VI) se puede cuantificar reteniendo el producto de su reacción con difenilcarbazida en una resina de intercambio catiónico [41]. Otra determinación donde se retiene el producto de reacción en una celda fluorimétrica empacada con una resina aniónica, es la reportada por #hen. y sus colaboradores para la determinación de c%thouanuros donde se utiliza una configuración de flujo continuo de confluencia de zonas para inyectar simultáneamente la muestra y el reactivo piridoxal 5-fosfato [42]. También para la determinación de aluminio [43] en agua Cañizares y Luque de Castro utilizan un sensor en flujo fluorimétrico donde el aluminio reacciona con la hidrazona del salicilpicolinoilaldehido (SAPH) para formar un complejo fluorescente que se retiene en C18 que esta empacado en la celda de flujo localizada en el paso óptico del instrumento. Utilizando la misma reacción pero una configuración diferente los mismos autores logran discriminar entre las diferentes especies de aluminio [44] (aluminio ácido reactivo, aluminio total monomérico, aluminio monomérico no lábil y lábil) por medio de una modificación al método Driscoll [45].
Existen otro tipo de sensores en flujo que se basan en la integración de la reacción y la detección por la inmovilización de uno o varios reactivos y/o el catalizador (que puede ser una enzima) en un soporte apropiado. La inmovilización de un reactivo Localizado en la celda de flujo se puede utilizar para la determinación de una gran variedad de especies por diferentes principios de detección. En un artículo reportado por Wang se explican algunos ejemplos representativos de este tipo de inmovilización utilizando electrodos con la superficie modificada e insertados en un sistema de flujo continuo [46]. En uno de estos sistemas, un electrodo de carbono es modificado inmovilizando Fe(III) / Fe(II) que responden amperométricamente a varios oxidos de nitrógeno [47]. Para la determinación de polifenoles en aceite de oliva se ha acoplado una extracción líquido-líquido en línea con un sensor localizado en la celda de flujo de un espectrofotómetro. El reactivo de Folin Cicalteau se inmoviliza en una resina aniónica y los fenoles son extraídos, con bicarbonato de sodio, realizando un cambio iterativo del flujo portador. Así los fenoles extraídos reaccionan con el reactivo inmovilizado para formar azul de molibdeno que también permanece retenido y se lee espectrofotométricamente. En este sensor se logra integrar la reacción derivatizadora, la retención y la detección in situ [48].
También se han desarrollado sensores bioquímicos basados en la detección directa del potencial de membrana inducido por una enzima inmovilizada que no requiere de la introducción de una referencia interna [49], o la inmovilización de la enzima ureasa o penicilasa en una membrana selectiva para el producto de reacción [50] sí como algunos biosensores potenciométricos y conductimétricos para la detección de pesticidas organofosforados [51]. Algunos de estos biosensores se basan en la inmovilización de una enzima en composites de grafito con una resina epóxica [52] pero pocos de los biosensores electroquímicos están acoplados a un sistema de flujo continuo debido a los problemas hidrodinámicos del sistema. Algunos de los electrodos en flujo se basan en el acoplamiento de un compuesto electroactivo con una gran resistencia mecánica para soportar el flujo continuo como es el composite de PVC-grafito [53, 54] y electrodos de grafito modificados por medio de enzimas inmovilizadas, como el desarrollado por Yarapolov y su grupo para la determinación de fenoles [55] o el biosensor para fosfatos utilizado por Gavalas y Chaniotakis [56].
Actualmente el Análisis por inyección en flujo esta enfocado al pretratamiento de muestras en línea para evitar la mayor manipulación de las mismas. Varios investigadores han logrado acoplar estos dos sistemas con gran éxito. Karlberg [57] y su grupo así como el grupo de Zhao-Lung Fang [58] han combinado el pretratamiento de muestras con técnicas de inyección en flujo y técnicas de inyección secuencial acopladas a electroforesis capilar.
Dentro del campo de análisis de alimentos donde el pre-tratamiento de la muestra suele ser laborioso y por lo tanto produce errores significativos en la cuantificación de los analitos, las determinaciones por FIA son una alternativa idónea. Noguira y su grupo [59] realizaron la determinación conductimétrica de cloruros en leche utilizando un sistema de inyección secuencial y una cámara de diálisis en la configuración FIA para eliminar interferencias de proteínas y sólidos suspendidos. En al análisis de vinos muchos parámetros se han automatizado disminuyendo el tiempo de análisis y de reactivos, por ejemplo: para la determinación selectiva de dióxido de sulfuro y ácido sulfhídrico se ha utilizado una membrana de separación para difusión de gases que elimina los pasos de destilación y titulación del método convencional [60]; para la determinación electroquímica simultánea de dióxido de azufre y ácido ascórbico [61] y en la determinación de azúcares reductores donde han utilizado un sistema automatizado de adiciones estándar y detección espectrofotométrica [62] y un sistema de inyección secuencial con una unidad de diálisis [63], en ambos métodos existe una ahorro apreciable de tiempo, entre 11 y 18 muestras por hora, y una precisión del 2 % aproximadamente.
Dentro del campo farmacéutico, Wei Li [64] desarrolló un sistema de análisis por inyección en flujo en la modalidad de "stopped-flow" para la determinación fluorimétrica simultanea de vitaminas B1, B2 y B6 en complejos vitamínicos realizando una disolución de las tabletas sólidas en línea; Almendral [65] y su grupo de trabajo han realizado una extracción líquido-líquido en línea de Bi(III) en productos para la gastritis y úlcera duodenal. La determinación se basa en la formación un complejo entre el tetrafeniarsenioso y el tetrayodobismutato (III), el producto de reacción se extrae con cloroformo utilizando una cámara de diálisis y se lee a 363 nm.
El análisis por inyección en flujo es una técnica tan versátil que se ha podido realizar estudios en línea y en vivo para la determinación de calcio en sangre insertando un tubo de microdiálisis en la oreja de un conejo obteniéndose una precisión del 3 % [66]. También se ha logrado producir, a nivel microescala, ácido maléico a partir de ácido fumárico el cual se aspiró a través de un sistema de flujo continuo donde se acoplaron reactores enzimáticos, con fumarasa y malato deshidrogenasa inmovilizadas, obteniéndose una conversión de hasta el 95 % y una precisión del 3 % [67].
Otra metodología bastante interesante debido a la configuración utilizada es la desarrollada por Danielsson y Valcárcel [68] donde evalúan la cantidad de nicotina en rapé utilizando membranas líquidas. La muestra sólida se pasa directamente, sin ningún pretratamiento, a través de la unidad de membrana líquida donde la nicotina pasa de la membrana (polipropilen-n-undecano) a un receptor ácido. Entre cada muestra la membrana se regenera añadiéndole n-undecano.
La tendencia de las metodologías por análisis por inyección en flujo es disminuir los pasos de pretratamiento de muestras y miniaturizar las metodologías de forma automatizada para que los errores de manipulación sean menores y aumente la precisión de los resultados, ya que es posible llevar a cabo varias determinaciones de una misma muestra en un tiempo menor de análisis lo que conlleva a una gran disminución de reactivos y por lo tanto de desechos, claves para mejorar el control de calidad de las mediciones analíticas. En un laboratorio de rutina, muchas veces, no es posible realizar las determinaciones por triplicado ya que se tienen que analizar muchas muestras y el costo de análisis sería muy elevado, lo que hace que el análisis por inyección en flujo sea la mejor alternativa en una gran cantidad de análisis. Existen varios trabajos que demuestran la disminución de tiempo y costos así como la alta precisión en las determinaciones por FIA. Por ejemplo, se ha realizado la determinación fluorescente de L-carnitina en preparados farmacéuticos obteniéndose una precisión del 0.96 % y una reproducibilidad para 144 muestras del 1.47 % [69].
Shiowatana [70] y su grupo diseñaron un sistema de flujo donde realizan una extracción secuencial para la especiación de metales en suelos, demostrando que el tiempo de extracción utilizando este sistema disminuía de sobre manera: de 4 a 5 horas en comparación con el método convencional que es de 2 a 3 días. En un trabajo realizado por Kolev y sus colaboradores demuestran que se obtiene una precisión adecuada, entre el 1 y 4 %, en la determinación amperométrica de fenoles en agua utilizando una cámara de pervaporación para eliminar la etapa de filtración o de extracción en fase sólida que se requiere para los métodos cromatogáficos [71].
En un estudio realizado para la determinación de nitratos y nitritos en agua se comparó el método convencional [72] con el método utilizado por FIA en cuanto a tiempo y costos por determinación [73]. En ambos métodos la determinación se basa en la reacción de los nitritos con la sulfanilamida para formar la sal de diazonio que reacciona con el N-1-naftiletilendiamina para medir un azo compuesto que se lee espectrofotométricamente a 540 nm. En el caso de los nitratos estos se reducen a nitritos por medio de una columna de cadmio. El ahorro que se logró utilizando la metodología por FIA fue del 85 % por determinación, con una frecuencia de muestreo de 37 determinaciones por hora contra 5 logradas por el método convencional.
Conclusiones
La mayoría de los programas de investigación, a nivel mundial, en Química Analítica están enfocados a mejorar la calidad en los resultados analíticos. El Análisis por Inyección en Flujo es una de las técnicas en la que este objetivo se puede cumplir y es una de las formas más sencillas para automatizar una determinación.
Dada su trascendencia en la calidad analítica, la automatización de las etapas del proceso analítico, que conectan la muestra con el instrumento, es una de las metas más importantes de la química analítica contemporánea. Sin embargo la automatización de estas etapas no es una tarea fácil. Por esta razón, es que aun continúan en uso metodologías manuales en la gran mayoría de los laboratorios analíticos de control.
Paralelamente se ha ido demostrando en los últimos años, que para la automatización de las etapas preliminares del proceso analítico, las técnicas continuas, particularmente el Análisis por Inyección en Flujo, es la forma más versátil, flexible y de menor costo para realizar una gran cantidad de operaciones del proceso analítico. Además de la adición de la muestra, dilución, calentamiento, mezclas, adición de reactivos (operaciones que igualmente se pueden realizar con analizadores discontinuos en discreto), con FIA se pueden realizar procesos de diálisis, pervaporación, destilación, extracción con disolvente y otras separaciones con un alto grado de precisión, exactitud y rapidez.
También es importante destacar que, además de involucrar costos muchísimo menores que los analizadores discontinuos en discreto, tienen mayor compatibilidad con diferentes detectores (potenciométricos, amperométricos, espectrofotométricos, AA, fluorescencia, etc).
Por lo tanto, se puede afirmar que la altísima relación eficiencia / costo que proporcionan las técnicas FIA para realizar procesos automáticos, lo hace altamente rentable y lo sitúa como una alternativa idónea para la obtención de resultados de gran calidad.
Referencias
1. Skeggs, L.T. Am. J. Pathol. 1957, 28, 311-315. [ Links ]
2. Ruzicka, J.; Hansen, E. H. Anal. Chim. Acta. 1975, 78, 145-157. [ Links ]
3. Nagy; Feher; Pungor, Anal. Chim. Acta. 1970, 52, 47-54. [ Links ]
4. White, V. R.; Filtzgerald, J. M. Anal Chem. 1975, 47, 903-908. [ Links ]
5. Stewart, K. K. Anal. Biochem. 1976, 70, 167-175. [ Links ]
6. Ruzicka, J.; Hansen, E. H.; Zagatto, E. A. Anal. Chim. Acta. 1977, 88, 1-16. [ Links ]
7. Painton, C. C.; Mottola, H. A. Anal. Chim. Acta 1983, 154, 1-16. [ Links ]
8. Painton, C. C.; Mottola, H. A. Anal. Chim. Acta 1984, 158, 67-76. [ Links ]
9. Ruzicka, J.; Hansen, E. H. Anal. Chim. Acta 1978, 99, 37-76. [ Links ]
10. Valcárcel, M.; Luque de Casto, M.D. Análisis por Inyección en Flujo, Monte de Piedad y Caja de Ahorros de Córdoba, España, 1984. [ Links ]
11. Painton, C.C.; Mottola, H.A. Anal. Chem. 1981, 53, 1713-1715. [ Links ]
12. Ruzicka, J.; Stewart, K. K. Anal. Chim. Acta. 1975, 79, 79-91. [ Links ]
13. Hansen, E.H.; Ruzicka, J. J. Chem. Ed. 1979, 56, 677-680. [ Links ]
14. Krug, F.J.; Bergamin Filho, H.; Zagatto, E.A.G.; Storgard-Jorgensen, S. Analyst 1977, 102, 503-505. [ Links ]
15. J. Mortatt, F. J.; Krug, L. C. R.; Pessenda; E. Zagatto, A. Analyst 1982, 107, 659-663. [ Links ]
16. Olsen, S.; Pessenda, L. C. R; Ruzicka J., Hansen, E. Analyst 1983, 108, 905-917. [ Links ]
17. Cañete, F.; Rios, A.; Luque de Castro, M. D.; Valcárcel, M. Analyst 1987, 112, 263-266. [ Links ]
18. Cañete, F.; Rios, A.; Luque de Castro, M. D., Valcárcel, M. Analyst 1987, 112, 267-270. [ Links ]
19. Fang, Z.; Ruzicka, J.; Hansen, E. Anal. Chim. Acta 1984, 164, 23-39.
20. Appeton, J. M. H.; Tyson, J. F. Anal. Chim. Acta 1986, 179, 269-278. [ Links ]
21. Chapin, T. P.; Johnson, K. S.; Coale, K. H. Anal. Chim. Acta 1991, 249, 469-478. [ Links ]
22. Edwards, R. T.; McKelvie, I. D.; Ferrett, P. C.; Hart, B. T.; Bapat, J. B. Anal. Chim. Acta 1992, 261, 287-294. [ Links ]
23. Liu, R. M.; Liu, D. J.; Sun, A. L. Analyst 1992, 117, 1767-1770. [ Links ]
24. Johnson, K. J.; Petty, R. L. Anal. Chim. Acta 1982, 54, 1185-1187. [ Links ]
25. Clarke, N.; Danielson, L. G.; Sparen, A. Inter. J. Environ. Anal. Chem. 1992, 48, 77-100. [ Links ]
26. Basson, W. D.; Van Staden, J. F. Water Research 1981, 15, 333-335. [ Links ]
27. Balconi, M. L.; Borgarello, M.; Ferraroli, R.; Realini, F. Anal. Chim. Acta 1992, 261, 295-299. [ Links ]
28. Rios, A.; Luque de Castro, M.D.; Valcárcel, M. Anal. Chem. 1985, 57, 1803-1809. [ Links ]
29. Cañete, F.; Rios, A.; Luque de Castro, M.D.; Valcárcel, M. Analyst 1987, 112, 263-266. [ Links ]
30. Olsen, S.; Pessenda, L. C. R.; Ruzicka, J.; Hansen, E. H. Analyst 1983, 108, 905-917. [ Links ]
31. Fang, Z.; Ruzicka, J.; Hansen, E. H., Anal. Chim. Acta 1984, 164, 23-39. [ Links ]
32. Gorton, L.; Ogren, L. Anal. Chim. Acta 1981, 130, 45-53. [ Links ]
33. Mascini, M.; Palleschi, G. Anal. Chim. Acta 1983, 145, 213-217. [ Links ]
34. Ruz, J.; Luque de Castro, M.D.; Valcárcel, M. Analyst 1987, 112, 259-261. [ Links ]
35. Beh, S.K.; Moody, G.J.; Thomas, J.D.R. Analyst 1991, 116, 459-462. [ Links ]
36. Xiaowen Wen; Kipton, H.; Powell, J.; Christian, G.D.; Ruzicka, J. Anal. Chim. Acta 1991, 249, 451-460. [ Links ]
37. Fernández, J. M.; Luque de Castro, M. D.; Valcárcel, M. Anal. Chim. Acta 1992, 264, 275-282. [ Links ]
38. Carlsen, M.; Christiansen, L.H.; Nielsen, J. Anal. Chim. Acta 1993, 274, 117-123. [ Links ]
39. Bryce, D. W.; Fernández Romero, J. M.; Luque de Castro, M.D. Anal. Lett. 1994, 27, 867-878. [ Links ]
40. Quiles, R.; Fernández Romero, J. M.; Fernández, E.; Luque de Castro, M. D.; Valcárcel, M. Clin. Chem. 1993, 39, 500-503. [ Links ]
41. Yoshimura, K. Analyst 1988, 113, 471-474. [ Links ]
42. Chen, D.; Luque de Castro, M.D.; Valcárcel, M. Talanta 1990, 37, 1049-1055. [ Links ]
43. Cañizares, P.; Luque de Castro, M.D. Anal. Letters 1994, 27, 247-262. [ Links ]
44. Cañizares, P.; Luque de Castro, M.D. Anal. Chim. Acta 1994, 295, 59-65. [ Links ]
45. Pereiro, M.R; Díaz García, M.E; Sanz-Medel, A. Analyst 1990, 115, 575-579. [ Links ]
46. Wang, J. Anal. Chim. Acta 1990, 234, 41-48. [ Links ]
47. Bonakdar, M.; YU, Y.; Mottola, H.A. Talanta 1989, 36, 219-225. [ Links ]
48. Cañizares, P.; Tena, M.T.; Luque de Castro, M.D. Anal. Chim. Acta 1996, 323, 55-62. [ Links ]
49. Ivnitskii, D.M.; Rishpon, J. Anal. Chim. Acta 1993, 282, 517-525. [ Links ]
50. Koncki, Robert; Walcerz, Isabela; Leszczynska, Ewa, J. Pharm. Biom. Anal. 1999, 19, 633-638. [ Links ]
51. Jaffrezic-Renault, N.; Martelet, C.; Clechet, P.; Nyamsi Soldatkin, A. P. Sens. Mater. 1996, 8, 161-167. [ Links ]
52. Céspedes, F.; Martínez-Fábregas, E.; Alegret, S. Anal. Chim. Acta 1993, 284, 21-26. [ Links ]
53. Albertús; Llerena; Alpízar; Cerdá; Luque; Ríos; Valcárcel, Anal. Chim. Acta 1997, 355, 23-32. [ Links ]
54. Luque, M.; Ríos, A.; Valcárcel, M., Anal. Chim. Acta 1999, 395, 217-223. [ Links ]
55. Yarapolov; Kharybin; Emnéus; Marko Varga; Gorton, Anal. Chim. Acta 1995, 308, 137-144. [ Links ]
56. Gavalas, Vasilis; Chaniotakis, Nicolás, Anal. Chim. Acta, 2001, 427, 271-277. [ Links ]
57. Kuban, P.; Engstrom, A.; Olsson, J.C.; Thorsen, G.; Tryzell, R.; Karlberg, B. Anal. Chim. Acta 1997, 337, 117-124. [ Links ]
58. Fanghan, Z.L.; Liu, Z.L.; Shen, Q. Anal. Chim. Acta 1997, 346, 135-143. [ Links ]
59. Silva Fernando V.; Souza, Gilberto B.; Feraz Luis, F.M.; Nogueira, Ana Rita A. Food Chem. 1999, 67, 317-322. [ Links ]
60. Mana, Hasson; Spohn, Uwe, Anal. Chem. 2001, 73, 3187-3192. [ Links ]
61. Cardwell, Terence; Christophersen, Melinda, Anal. Chim. Acta, 2000, 416, 105-110. [ Links ]
62. Cañizares Macías, M. P.; Hernández Garciadiego, L.; Gómez Ruíz H. J. Food Sci. 2001, 66, 407-411. [ Links ]
63. Araujo, Alberto N.; Lima, José L.; Rangel, António; Segundo, Marcela, Talanta 2000, 52, 59-66. [ Links ]
64. Wei, Li; Jian, Chen; Bingren, Xian; Dengkui, Anal. Chim. Acta 2000, 408, 39-47. [ Links ]
65. Alonso, A.; Almendral, M.J.; Baez, M.D.; Porras, M.J.; López Lavín, F.; García de María, C. Anal. Chim. Acta 2000, 408, 129-133. [ Links ]
66. Yuming, Huang; Zhujun, Zhang; Jiagen, Lv; Hua, Cheng, Anal. Chim. Acta 2000, 419, 175-184. [ Links ]
67. Almuaibed Ala'ddin, M. Anal. Chim. Acta 2001, 428, 1-6. [ Links ]
68. Luque Pérez, E.; Ríos, A.; Valcárcel, M., Danielsson, L.G.; Ingman, F. Anal. Chim. Acta 1999, 387, 155-164. [ Links ]
69. Manjón, A.; Obón, J. M., Iborra, J. L.; Anal. Biochem., 2000, 281, 176-181. [ Links ]
70. Shiowatana, J.; Tantidanai, N.; Nookabkaew, S.; Nacapricha, D. Environ. Int. 2001, 26, 381-387. [ Links ]
71. Sheikheldin, Sami; Cardwell, Terence; Cattrall, Robert; Luque de Castro, M. D.; Kolev, Spas, Anal. Chim. Acta 2000, 419, 9-16. [ Links ]
72. Annual Book of ASTM Standards. Water and Enviromental Technology, Standard Test Methods for Nitrite and Nitrate in water, Vol. 11.01, D3867-90, 1991. [ Links ]
73. Cabrera Molina, Erika; Cañizares Macías, M. P., Desarrollo de un método automatizado para la determinación de nitratos y nitritos en aguas por FIA, Tesis de Licenciatura, Facultad de Química, UNAM, 1999. [ Links ]

