










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.46 no.2 Ciudad de México abr./jun. 2002
Investigación
Efecto de la (E)-3-butil-8-estiri1-1-propilxantina sobre la asimetría motora en rata
Alejandro Muñoz-Zurita, Jesús Sandoval-Ramírez,* Carmen Parra-Cid y Daniel Limón-Pérez de León
Facultad de Ciencias Químicas. Benemérita Universidad Autónoma de Puebla. México. Apartado postal 1742, Puebla 72000, Pue. México. Tel.: 01-222-2295-500 Ext. 7382; Fax: 222-2295-584. E-mail: jsandova@siu.buap.mx
Recibido el 9 de febrero del 2002.
Aceptado el 11 junio del 2002.
Resumen
Se describe la síntesis de la (E)-3-butil-8-estiri1-1-propil-xantina (A15Bu) a partir de 6-aminouracilo (6) y un estudio biológico con el modelo experimental antiparkinsoniano de conducta de giro, en ratas de cepa Wistar. Al administrar el compuesto A15Bu en ratas lesionadas con 6-hidroxidopamina (6-OHDA) en la sustancia nigra pars compacta la asimetría motora se redujo en un 75 %.
Palabras clave: Antagonistas de receptores de adenosina, xantinas, quimioselectividad, agentes antiparkisonianos, modelo de asimetría motora.
Abstract
Herein we describe the synthesis of (E)-3-butyl-1-propyl-8-styrylxanthine (A15Bu) starting from 6-aminouracil (6) and a biological study with the antiparkinsonian experimental model of rotational behavior using Wistar rats. When the xanthine derivative A15Bu was administered to rats lesioned with 6-hydroxydopamine (6-OHDA) at the substantia nigra pars compacta, the asymmetric motor behavior was reduced by 75 %.
Keywords: Adenosine receptors antagonist, xanthines, chemoselectivity, antiparkinsonian drugs, asymmetric motor model.
Introducción
La adenosina es una molécula producida en algunos órganos y tejidos que modula una gran variedad de funciones biológicas en el sistema nervioso central (SNC) y periférico, su efecto se lleva a cabo a través de la interacción con uno o más de los cuatro subtipos de receptores que se conocen en la actualidad: A1, A2A, A2B y A3 [1]. Se ha propuesto que los subtipos A1 y A2A juegan papeles complementarios en la regulación de la adenosina [2]. El subtipo A2B se expresa en células de tejidos dañados o inflamados y facilita la movilización del calcio intracelular [2]. Las funciones del subtipo A3 no se han caracterizado totalmente pero los compuestos que activan o inhiben a estos receptores son estudiados por su potencial uso terapéutico en enfermedades del corazón y el cáncer [3].
Las metilxantinas como la teofilina (1) son conocidas desde hace mucho tiempo por sus efectos estimulantes sobre el SNC debido a su efecto bloqueador de los receptores de adenosina, aunque no son antagonistas selectivos; sin embargo, se ha reportado que los antagonistas de los receptores adenosinérgicos del subtipo A2A pueden ser útiles como agentes antiparkinsonianos; no obstante, se desconocen muchos aspectos del papel que desempeñan en dicho padecimiento [4].
Se ha descubierto un gran número de antagonistas A2 muy selectivos entre los que se incluyen por ejemplo, derivados de xantinas, adeninas, 1,2,4-triazolo[1,5-a]quinoxalinas y pirazolo[3,4-d]pirimidinas como 2, 3, 4 y 5, respectivamente (Fig. 1).
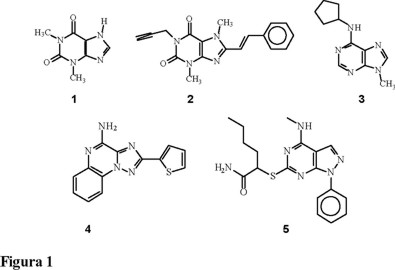
Los compuestos derivados de la teofilina (1) que contienen en la posición 1 a grupos lipofílicos como el n-propilo son reconocidos fácilmente por los receptores de la adenosina [5]. Además, se ha descubierto que el sustituyente estirilo con isomería (E) en la posición 8 confiere selectividad por los receptores adenosinérgicos del tipo A2A [6]. Por otra parte, recientemente se ha reportado la participación de los receptores A2A en las conductas motoras [7] y se ha encontrado que los nuevos antagonistas A2A revierten los estados catalépticos inducidos por fármacos antagonistas dopaminérgicos D1 y D2 cuando son aplicados en el caudoputamen [7]; dicha actividad también se observa cuando disminuye la producción de dopamina [8]. Esto sugiere que los receptores A2A modulan la activación de los receptores D2 regulando la transmisión GABA-érgica estriadopalidal [9]. Otros estudios han demostrado que el efecto estimulante de los antagonistas A2A, se debe a un bloqueo de los receptores A2A en el estriado [10]. Se sugiere que los antagonistas adenosinérgicos de tipo A2A tienen efectos motores y pueden revertir la asimetría motora o el estado hipocinético; inclusive, se ha reportado recientemente que nuevos antagonistas A2A se encuentran ya en la fase II de experimentación [11]. Nosotros nos hemos interesado en la síntesis de xantinas como probables antagonistas de receptores de la adenosina selectivos, con efectos sobre la conducta motora y por tanto, como agentes antiparkinsonianos que permitan una nueva terapéutica contra esta enfermedad neurodegenerativa. La síntesis de la molécula modelo se llevó a cabo tomando como base la experiencia lograda en la síntesis de otros derivados xantínicos [12, 13].
Resultados y discusión
La síntesis de la xantina A15Bu funcionalizada en posiciones 1-, 3-, y 8- se llevó a cabo a partir del 6-aminouracilo (6) de acuerdo con el esquema de trabajo mostrado en la Fig. 2 [12].
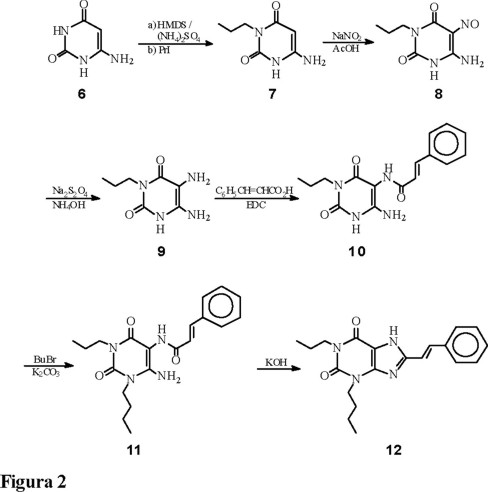
Para llevar a cabo la alquilación selectiva de 6 en posición 3, se forma in situ el derivado trisililado utilizando cantidades prácticamente equimolares de hexametildisilazano (HMDS) y cantidades catalíticas de (NH4)2SO4 seguido por la adición de un equivalente de yoduro de n-propilo. El derivado alquilado 7 se aisla por precipitación en agua, con un 92 % de rendimiento en ausencia de producto dialquilado.
La nitrosación quimio- y regioselectiva en posición 5, para obtener el derivado 8 se realiza utilizando nitrito de sodio en medio ácido acuoso. Nosotros hemos encontrado que los mejores rendimientos (aprox. 80 %) se obtienen cuando la reacción se realiza a temperatura ambiente y dichos rendimientos son equiparables o superiores a los reportados en la literatura [14]. Los derivados nitroso son fácilmente reconocibles por su intenso color naranja y por la banda intensa en el infrarrojo hacia 1510 cm-1 [15].
Para realizar la reducción del grupo nitroso y obtener el compuesto 9, se empleó el método que utiliza ditionito de sodio en medio básico. Dado que la diamina 9 es muy inestable, ésta se debe someter de forma inmediata a la próxima reacción en ausencia de luz y oxígeno, para evitar la descomposición. Con lo anterior la diamina 9 se obtiene con un 65 % de rendimiento.
La obtención de la amida 10 se efectuó haciendo reaccionar el ácido trans-cinámico con la diamina 9 y empleando el clorhidrato de 1-[3-(dimetilamino)propil]-3-etilcarbodiimida (EDC) como agente acoplante, con rendimientos cuantitativos. Esta es una reacción totalmente quimioselectiva pues solamente reacciona el grupo amino unido a la posición 5.
Los datos espectroscópicos de IR y de RMN de protón establecen que la doble ligadura del grupo estirilo permanece inalterada, pues tanto el dato de 971 cm-1 como la constante de acoplamiento de 15.8 Hz son características para una doble ligadura con estereoquímica trans [15, 16].
Sobre la amida 10 se puede realizar una alquilación quimioselectiva en posición 1 utilizando un equivalente de bromuro de n-butilo, en presencia de K2CO3, obteniéndose un rendimiento del 76 %.
Finalmente, la reacción de ciclización para obtener la (E)-3-butil-8-estiri1-1-propilxantina se llevó a cabo por tratamiento de 11 con una solución acuosa de KOH, en metanol, a reflujo durante 3 h, con un rendimiento del 69 %.
En la literatura, para caracterizar a compuestos uracílicos o xantínicos se han utilizado por lo general, los métodos espectroscópicos de RMN, IR y Masas haciendo caso omiso de la espectroscopía UV-Vis. En este artículo presentamos los datos de UV-Vis de los derivados uracílicos 7, 8, 10 y 11 así como el de la xantina A15Bu que al igual a los obtenidos de otros derivados en nuestro laboratorio muestran una gran utilidad en la elucidación de sus estructuras; así, los derivados del 6-aminouracilo con el grupo (E)-estirilcarboxamido unido a la posición 5 generan espectros con λmaxpromedio de 270 nm, en tanto que el derivado xantínico A15Bu muestra una absorción en 342 nm.
Farmacología
Para llevar a cabo la evaluación de la xantina A15Bu, se utilizaron ratas macho adultas de la cepa Wistar, provenientes del Bioterio Claude Bernard de la BUAP, con un peso de 250-280 g los cuales fueron mantenidos en cajas de lámina acrílica las cuales fueron mantenidas bajo las mismas condiciones de agua y alimento, con un ciclo de luz / oscuridad de 12 × 12 h, a una temperatura de 22 °C.
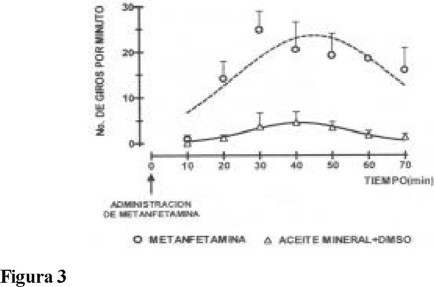
La evaluación de conducta motora se realizó a través del Modelo de conducta de giro: a un grupo de animales se les administró 16 µg de 6-OHDA (disueltos en una solución acuosa de ácido ascórbico al 0.1 %) en la sustancia nigra pars compacta mediante una cirugía estereotáxica en las coordenadas: AP = -4.9; L = +2.0; P = -6.5 [17]. Ocho días después se evaluó la lesión dopaminérgica administrando metanfetamina (8 mg / kg vía s.c.). Se consideró el número de giros ipsilaterales para cada roedor a partir de la administración de metanfetamina 70 min. Solamente 16 animales presentaron diez o más giros por min (Fig. 3).
Quince días después de la lesión, los animales fueron divididos en el grupo control (n = 8) al cual se le administró aceite mineral en dimetilsulfóxido (DMSO) en una relación 1:5 y al grupo experimental (n = 8) se le administró el A15Bu como probable antagonista A2A a una dosis de 1.0 mg / kg vía s.c. Después de diez minutos a ambos grupos nuevamente se les realizó la prueba de conducta de giro con metanfetamina (8.0 mg / kg vía s.c.) y se evaluó el número de giros por 70 min (Fig. 4).

En la Fig. 3 se muestra la evaluación de la lesión dopaminérgica en la cual se contó el número de giros ipsilaterales entre el minuto 10 al 70 después de la administración de la metanfetamina (8 mg / kg vía s.c.) a ambos grupos de ratas. Se observó un incremento significativo del número de giros a partir del minuto 20 hasta alcanzar un máximo en el número de giros entre el minuto 40 al 70, respecto al grupo control. El número máximo de giros fue de 20.45 ± 0.9 al minuto 40 y de 18.45 ± 1.0 al minuto 60 de la administración de la metanfetamina. El grupo al que se le administró el vehículo durante los 70 minutos de registro (triángulos, Fig. 3) no presentó giros ipsilaterales.
Por lo tanto los siguientes experimentos para evaluar el posible antagonista se realizaron entre el minuto 40 al 70 después de la administración de metanfetamina. Cinco días después y bajo las mismas condiciones experimentales, se administró el derivado de las xantinas A15Bu como probable antagonista A2A a razón de 1 mg / kg vía s.c. al grupo experimental y al grupo control el vehículo. La administración del vehículo se hizo diez minutos antes de la administración de la metanfetamina (8 mg / kg vía s.c.).
Encontramos que hubo una disminución del número de giros aunque no significativa en el minuto cuarenta (Fig. 3). Sin embargo, encontramos una diferencia significativa *p < 0.05 del 40 al minuto 70 de experimentación. El decremento significativo en el número de giros se inició a partir del minuto cincuenta y esta diferencia se mantuvo hasta el minuto 70, en la cual hubo una disminución significativa de un 75 % respecto a su grupo control (Fig. 4).
Así, en este estudio mostramos que la asimetría motora producida por el daño dopaminérgico con 6-OHDA en la sustancia nigra pars compacta (Fig. 4) fue revertida cuando se aplicó el compuesto A15Bu como supuesto antagonista A2A.
Parte experimental
Los puntos de fusión fueron determinados por la técnica de capilar abierto en un fusiómetro Melt-temp usando la escala de grados centígrados y no fueron corregidos. Los espectros de infrarrojo se midieron en un espectrofotómetro NICOLET-MAGNA FT-IR-750, los datos se expresan en cm-1. Los espectros de UV fueron obtenidos en un equipo BECKMANN DU-7000 empleando soluciones etanólicas al 98 %; las longitudes de onda se expresan en nm. Los espectros de RMN de 1H y 13C se realizaron en un espectrómetro JEOL eclipse a 400 y 100 MHz, respectivamente, utilizando DMSO-d6 y CDCl3 como disolventes y las señales de los protones remanentes de los disolventes deuterados como referencia interna. Los espectros de masas se determinaron a 70 eV en un espectrómetro Hewlett Packard 5989A, acoplado a un cromatógrafo de gases Hewlett Packard 5990 serie II; los datos se expresan en relación m/z.
6-amino-3-propiluracilo (7). En un matraz de fondo redondo de 50 mL, provisto de refrigerante para reflujo, se prepara una suspensión de 5.08 g (40 mmol) de 6-aminouracilo, en 12.65 ml (60 mmol) de HMDS y 0.243 g (1.92 mmol) de (NH4)2 SO4; la mezcla se calienta bajo atmósfera de argón y agitación magnética hasta llegar al reflujo con lo cual se obtiene una solución transparente y homogénea; después de 15 minutos se disminuye la temperatura (40-50 °C), se añaden 3.9 mL (40 mmol) de yoduro de n-propilo y el medio de reacción se calienta hasta reflujo. La reacción se monitorea por CCF usando como eluyentes CH2Cl2: CH3OH (9:1) hasta la desaparición de la materia prima (aprox. 3 h). Se deja enfriar hasta 40 °C y se añaden 20 mL de solución saturada de Na2S2O3 (45g / 100 mL) con lo cual se obtiene una suspensión que se agita durante 30 minutos y a continuación se agregan lentamente 20 mL de solución saturada de NaHCO3. Se filtra al vacío, se enjuaga con agua fría, benceno y finalmente con éter dietílico. Rendimiento: 92 %. Polvo blanco amorfo: pf 267-269 °C (lit. 275 °C [14]); UV (EtOH) λmax (ε) 261 (3508) nm; IR (KBr) νmax 2930, 1741, 1631 cm-1; RMN 1H (DMSO-d6, 400 MHz) δ 6.32 (2H, b, -NH2), 4.53 (1H, s, H-5), 3.59 (2H, t, -CH2-N-3, J=7.32 Hz), 1.46 (2H, m, -CH2CH2CH3, 7.32 Hz), 0.80, (3H, t, -CH2CH2CH3, J=7.32 Hz); RMN 13C (DMSO-d6, 100 MHz) δ 163.67 (C-4), 154.53 (C-2), 151.92 (C-6), 74.66 (C-5), 21.52 (-CH2CH2CH3), 11.73 (-CH2CH2CH3); EMIE: m/z (int. rel.): 169 [M]+ (22), 127 (100).
6-amino-5-nitroso-3-propiluracilo (8). En un matraz de 50 ml equipado con embudo de adición, barra magnética y atmósfera inerte se disuelve 7 (5 mmol, 0.84 g) en AcOH al 80 %, se comienza a agregar 10 mmol (0.69 g) de NaNO2 disuelto en 5 mL de agua durante un lapso de 45 minutos. Al caer las primeras gotas, la solución cambia de color (de blanquecina a violácea) y conforme se adiciona mas solución de NaNO2, aparece un sobrenadante naranja o amarillo intenso. Al finalizar la adición de la solución de NaNO2 se deja en agitación vigorosa durante 40 minutos más y se enfría a 0 °C para que el producto precipite. Se filtra al vacío y se enjuaga con agua fría. Se seca en un desecador al alto vacío. Rendimiento: 79 %. Polvo amarillo amorfo: pf > 300 °C; UV (EtOH) λmax (ε) 320 (5920) nm; IR (KBr) νmax 3294, 3185, 1511 cm-1; RMN 1H (DMSO-d6, 400 MHz) δ 11.37 (1H, b, N1-H), 3.79 (2H, t, -CH2-N-3, J=7.32), 1.60 (2H, m, -CH2CH2CH3, J=7.32), 0.89 (3H, t, -CH2CH2CH3, J= 7.32); RMN 13C (DMSO-d6, 100 MHz): δ 161.84 (C-4), 150.27 (C-2), 145.49 (C-6), 140.31 (C-5), 40.23 (-CH2-N-3), 21.37 (-CH2CH2CH3), 11.76 (-CH2CH2CH3).
5,6-diamino-3-propiluracilo (9). En un matraz de 100 mL provisto con barra de agitación y canasta de calentamiento, se prepara una solución de 10 mmol (1.98 g) de 6-amino-5-nitroso-3-propiluracilo (8) en 50 mL de NH4OH al 12.5 % acuoso, la solución color rojo intenso se calienta hasta que alcance una temperatura cercana a los 70 °C, enseguida se comienza a agregar el Na2S2O4 (aproximadamente 3 equivalentes, 0.522 g) en pequeñas porciones durante un lapso de 10 minutos. La cantidad que se agrega debe ser suficiente para que la solución se decolore. Inmediatamente se destila el agua al vacío hasta que queden aproximadamente 15 mL de agua, con lo cual se forma un precipitado cremoso, que se filtra rápidamente al vacío y se somete a la próxima reacción debido a su inestabilidad ante el ambiente. Rendimiento: 60-65 %. Agujas cristalinas amarillas, descomponen antes de fundir.
(E)-6-amino-5-estirilcarboxamido-3-propiluracilo (10). En un matraz de fondo redondo, provisto de barra de agitación y atmósfera inerte se prepara una suspensión de 6.5 mmol (1.19 g) de 5,6-diamino-3-propiluracilo (9), 6.5 mmol (1.18 g) de ácido trans-cinámico y 6.5 mmol (1.2 g) de EDC en 60 mL de solución al 50 % de CH3OH y se deja en agitación constante. La reacción se sigue por placa (CH2Cl2: CH3OH, 9:1) hasta que se consuma la materia prima (aprox. 10 h). Se enfría la suspensión a 0 °C y el precipitado formado se filtra al vacío dentro de la campana de extracción. El precipitado se enjuaga con agua fría y después con CH3OH helado. Se seca al alto vacío. Rendimiento: 76.9 %. Polvo blanco amorfo: descompone antes de fundir; UV (EtOH) λmax (ε) 272 (6670) nm; IR (KBr) νmax 3318, 3187, 1648 cm-1; RMN 1H ( DMSO-d6, 400 MHz): δ 8.7 (1H, b, NH amida), 7.41 (6H, m, 5 arom. y -CH=), 6.85 (1H, d, -CH=, J=16.08), 6.11 (2H, b, -NH2), 3.6 (2H, t, -CH2-N-3, J= 6.2), 1.5 (2H, m, -CH2CH2CH3, J= 7.32), 0.81 (3H, t, -CH2CH2CH3, J= 7.32); RMN 13C (DMSO-d6, 100 MHz): δ 165.58 (C=O amida), 161.15 (C-4), 150.49 (C-6), 150.39 (C-2), 139.44(-CH=), 135.50 (arom.), 130.04 (arom.), 129.55 (arom.), 128.04 (arom.), 122.93 (=CH-), 87.69 (C-5), 40.20 (-CH2-N-3), 21.50 (-CH2CH2CH3), 11.76 (-CH2CH2CH3).
(E)- 6-amino-1-butil-5-estirilcarboxamido-3-propiluracilo (11). En un matraz de fondo redondo de 50 mL, provisto con barra de agitación y atmósfera inerte, se disuelve 1 mmol (0.314 g) de 10 en 5 mL de DMF, a la solución se agrega 1 mmol (0.138 g) de K2CO3 y posteriormente se añaden 1.5 mmol de CH3(CH2)3Br y se deja en agitación constante; la reacción que se va siguiendo por CCF (CH2Cl2: CH3OH, 9:1), hasta que se consuma el material de partida. Se agregan 8 mL de agua destilada y se forma un precipitado blanquecino que se enfría a 0 °C y posteriormente se filtra al vacío. Se purifica al disolverse en 5 mL de DMF y precipitando por la adición de 10 mL de agua, se filtra al vacío y se seca adicionando 15 mL de éter dietílico. Rendimiento: 66 %. Polvo blanco amorfo: pf 273-274 °C; UV (EtOH) λmax (ε) 271 (8523) nm; IR (KBr) νmax 3319, 1731, 1652 cm-1; RMN 1H (CDCl3, 400 MHz): δ 8.14 (1H, b, -NH amida), 7.30 (5H, m, arom.), 6.76 y 7.55 (1H, d, -CH=, J=15.36), 5.82 (2H, b, -NH2), 3.78 (4H, m, -CH2-N-1 y -CH2-N-3, J=7.32), 1.58 (4H, m, -CH2CH2CH3, y -CH2CH2CH2CH3, J=7.32), 1.36 (2H, m, -CH2CH2CH2CH3, J=7.32), 0.91 (3H, t, -CH2CH2CH3, J=7.32), 0.88 (3H, t, -CH2CH2CH2CH3, J=7.32); RMN 13C (CDCl3, 100 MHz) δ 165.89 (C=O amida), 160.13 (C-4), 150.06 (C-6), 148.52 (C-2), 142.17 (arom.), 134.41 (-CH=), 130.06 (arom.), 129.39 (arom.), 128.90 (arom.), 128.03 (arom.), 119.88 (=CH-), 92.05 (C-5), 43.43 (-CH2-N-3), 43.20 (-CH2-N-1), 30.11 (-CH 2CH2CH2CH3), 21.20 (-CH2CH2CH3), 20.09 (-CH2CH2CH2CH3), 13.75 (-CH2CH2CH3), 11.37 (-CH2CH2CH2CH3).
(E)-3-butil-8-estiri1-1-propilxantina (12). Se solubiliza 1 mmol (0.369 g) de 11 en 4 mL de solución de KOH al 20 %, se adicionan 30 mL de metanol, se calienta hasta reflujo durante 3 horas, la reacción se monitorea por CCF (CH2Cl2: CH3OH, 9:1). Cuando se ha consumido la materia prima se adiciona lentamente HCl conc. hasta que el pH ~3. Se enfría para precipitar el producto, posteriormente se filtra al vacío y se adicionan 15 mL de agua fría, finalmente se seca adicionando 15 mL de éter dietílico. Rendimiento 69 %. Polvo blanco amorfo: pf 278-280 °C; UV (EtOH) λmax (ε) 342 (8785) nm; IR (KBr) νmax 3388, 1705, 1662, 1642 cm-1; RMN 1H (CDCl3, 400 MHz): δ 13.20 (1H, b, N-H), 7.36 (5H, m, arom.), 7.09 y 7.77 (1H, d, -CH=, J= 15.36), 4.19 (2H, t, -CH2-N-3, J=7.32), 4.11 (2H, t, -CH2-N-1, J=7.32), 1.80 (4H, m, 2 -CH2CH2CH3 y -CH2CH2CH2CH3, J=7.32), 1.43 (2H, m, -CH2CH2CH2CH3, J=7.32), 0.99 (6H, m, -CH2CH2CH3 Y CH2CH2CH2CH3, J=7.32); RMN 13C (CDCl3, 100 MHz): δ 156.00 (C-6), 151.40 (C-2 ), 150.98 (C-8), 149.9 (C-4), 137.03 (=C-), 135.71 (arom.), 129.39 (arom.), 128.98 (arom.), 127.28 (arom.), 115.39 (-C=), 107.37 (C-5), 43.83 (-CH2-N-3), 43.61 (-CH2-N-1), 30.16 (-CH2CH2CH2CH3), 21.5 (-CH2CH2CH3), 20.11(-CH2CH2CH2CH3), 13.88(-CH2CH2CH3), 11.51 (CH2CH2CH2CH3); EMIE: m/z (int. rel.): 352 [M]+ (100).
Agradecimientos
A.M.-Z. agradece al Consejo Nacional de Ciencia y Tecnología el apoyo económico otorgado a través de la beca crédito 121993.
Bibliografía
1. Hess, S.; Müller, C. E.; Frobenius, W.; Reith, U.; Klotz, K. N.; Eger, K. J. Med. Chem. 2000, 43, 4636-4646. [ Links ]
2. Poulsen, S. A.; Quinn, R. J. Bioorg. & Med. Chem. 1998, 6, 619-641. [ Links ]
3. Borman, S. Chem. & Eng. News. 2001, 79, 37-40. [ Links ]
4. Müller, C. E. Drugs of the Future. 2000, 25, 1043-1052. [ Links ]
5. Daly, J. W.; Padgett, W. L.; Shamin, M. T. J. Med. Chem. 1986, 29, 1305-1308. [ Links ]
6. Shimada, J.; Koike, N.; Nonaka, H.; Shiozaki, S.; Yanagawa, K.; Kanda, T.; Kobayashi, H.; Ichimura, M.; Nakamura, J.; Kase, H.; Suzuki, F. Bioorg. Med. Chem. Lett. 1997, 7, 2349-2352. [ Links ]
7. Hauber, N. J.; Sauer, R.; Müller, C. E. Neuroreport. 1998, 9, 1803-1806. [ Links ]
8. Fenu, S.; Pinna, A.; Ongini, E.; Morelli, M. Eur. J. Pharmacol. 1997, 321, 143-147. [ Links ]
9. Gonzalez, B.; Galvan, A.; Floran, L.; Aceves, J.; Floran, B. Meeting of Neuroscience. Miami Beach, Fl. 1999, Abstr. No. 468.2. [ Links ]
10. Floran, B.; Sierra, A.; Floran, L.; Aceves, J. Meeting of Neuroscience. Miami Beach, Fl. 1999, Abstr. No. 468.3. [ Links ]
11. Przuntek, H. J. Neurol. 2000, 247, 19-24. [ Links ]
12. Müller, C. E., Sandoval-Ramírez, J. Synthesis. 1995, 1295-1299. [ Links ]
13. Hayallah, A. M.; Sandoval-Ramírez, J.; Reith, U.; Schobert, U.; Preiss, B.; Schumacher, B.; Daly, J. W.; Müller, C. E. J. Med. Chem. 2002, 47, 1500-1510. [ Links ]
14. Müller, C. E. Synthesis 1993, 125-128. [ Links ]
15. Nakanishi, K.; Solomon, P. H. in "Infrared Absorption Spectroscopy". Holden-Day Inc. New York, 1977, 45. [ Links ]
16. Friebolin, H. in "Basic One- and Two-Dimensional NMR Spectroscopy". VCH Publ. New York, 1993, 87. [ Links ]
17. Paxinos, G., Watson, Ch. in "The Rat Brain in Stereotaxic Coordinates". Academic Press. San Diego, 1998, 44. [ Links ]

