










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.45 no.4 Ciudad de México oct./dic. 2001
Investigación
Highly Diastereoselective Addition of a Racemic β-Alanine Enolate Derivative to Electrophiles
Jaime Escalante,*,1 Ana Lilia Hernández,1 and Eusebio Juaristi*,2
1 Centro de Investigaciones Químicas, Universidad Autónoma del Estado de Morelos, Av. Universidad 1001, Cuernavaca, Mor., 62210, México. E-mail: escalante_jaime@hotmail.com
2 Departamento de Química, Centro de Investigación y de Estudios Avanzados del IPN, Apartado Postal 14-740, México 07000, D.F. E-mail: juaristi@relaq.mx
Recibido el 15 de noviembre del 2001.
Aceptado el 11 de diciembre del 2001.
Dedicated to Professor Fernando Walls, Instituto de Química, UNAM, on the occasion of his 70th birthday.
Abstract
β-Alanine, an inexpensive β-amino acid, was converted into the 2-phenylperhydropyrimidin-4-one derivative (1), which can be alkylated with high diastereoselectivity via the corresponding enolate. The high stereoselectivity observed for the reaction of (1)-Li with electrophiles seems to be due to steric hindrance generated by an axial disposition of the phenyl group at C(2), which directs addition from the enolate face opposite to this group. These results pave the road to the enantioselective synthesis of α-substituted β-amino acids.
Keywords: β-amino acid, diastereoselective alkylation, perhydropyrimidinone.
Resumen
β-Alanina, un aminoácido accesible, fue transformado al derivado 2-fenilperhidropirimidín-4-ona (1), que puede ser alquilado con alta diastereoselectividad mediante el enolato correspondiente. La alta estereoselectividad observada para la reacción de (1)-Li con electrófilos parece ser debida al impedimento generado por la disposición axial del grupo fenilo en C(2), el cual dirige la adición por la parte opuesta a este grupo. Estos resultados facilitan el camino a la síntesis enantioselectiva de β-aminoácidos substituidos en α.
Palabras clave: β-aminoácidos, alquilación diastereoselectiva, per-hidropirimidinona.
Introduction
Perhydropyrimidin-4-ones (I) are interesting heterocyclic compounds that represent protected forms of β-amino acids and are chiral precursors for the asymmetric synthesis of α-substituted β-amino acids [1]. The presence of the cyclic amidal moiety allows the acid hydrolysis of these compounds, affording directly the corresponding β-amino acids (Scheme 1).
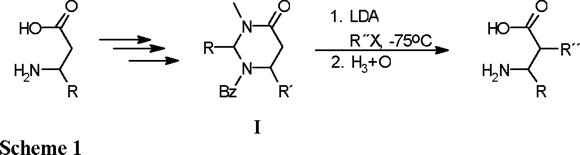
The preparation of β-amino acids is an interesting topic that now has attracted a great deal of attention, because unusual amino acids, such as α-substituted α-amino acids, β-amino acids, and γ-amino acids, allow the preparation of novel types of peptides that are catalysts or carriers of biologically active residues with pharmacologically interesting properties [2]. In order to design new enzyme-type ligands, conformational constraints have been introduced in the peptide chain, and this approach has provided an important rationalization for protein ligand development. In this context, the incorporation of unusual amino acids [3] results in conformational restriction and increased rigidity, leading to enhanced resistance towards protease enzymes and to the favoring of a particular secondary structure.
With the aim of obtaining α-substituted β-amino acids in enantiomerically pure form, the functionalization at C(5) of the perhydropyrimidin-4-ones through alkylation of the corresponding enolates has been designed by Juaristi [4], Cardillo [5] and Konopelski [6]. The presence in the heterocyclic ring of one or more stereocenters favors high diastereoselection in the alkylation step (Scheme 2).
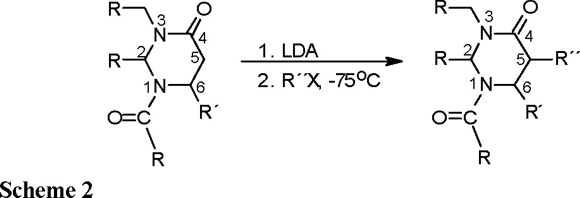
In particular, in view of the successful development of 2-t-butylperhydropyrimidinones for the preparation of α-alkyl β-amino acids [4], it was considered that the 2-phenylperhydropyrimidin-4-one derivative (1) might serve as an effective chiral substrate for the synthesis of α-substituted β-amino acids.
Results and discussion
A. Synthesis of 1-benzoyl-2-phenyl-3-methylperhydropyrimidin-4-one (1). The heterocycle rac-(1) was prepared from β-alanine by initial conversion to its methyl ester (2) and then to the corresponding N-methylamide (3), which formed a Schiff base with benzaldehyde (azeotropic removal of water). Cyclization of imine (4) with benzoyl chloride/DMAP afforded the desired rac-perhydropyrimidinone (1), with an overall yield of 51 % (Scheme 3).

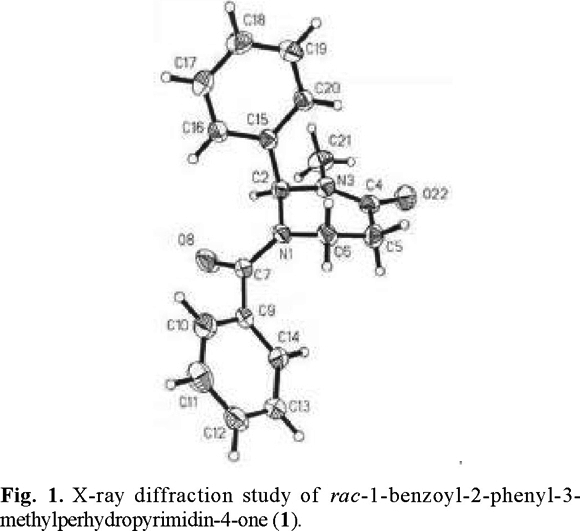
B. X-Ray diffraction study of 1-benzoyl-2-phenyl-3-methylperhydropyrimidin-4-one (1). Because of the present interest in the understanding of the precise structure of nitrogen-containing heterocycles [4d], and because such information can be important in ascertaining the factors responsible for the stereoselectivities observed (see below), we carried out X-ray structural analysis with a suitable crystal of perhydropyrimidinone (1). A view of the solid-state structure of (1) is provided in figure 1. The pyrimidinone ring is rather flat and has a sofa conformation with four of the six atoms approximately in a plane, and the nitrogen N(1) and carbon C(6) out of plane. The most interesting feature of the crystal structure is, however, that the six-membered ring adopts a conformation with an axial phenyl group, just like the tert-butyl group in the 2-t-butyl analogs. [4]. The practical consequences are also significant: if the enolate would still have a conformation with an axial phenyl group, one of its faces would be sterically hindered for attack by electrophiles. This was indeed the case, as described in the following section.
C. Diastereoselectivity of alkylation of enolate (1)-Li. The alkylation products 5, 7 and 9 were formed by treatment of enolate 1-Li, generated with lithium diisopropylamide (LDA) in THF, with halides RX at − 78 °C. High diastereoselectivity (ds > 96 %) was found as indicated by integration of the 13C NMR spectra of the crude products (Table 1).

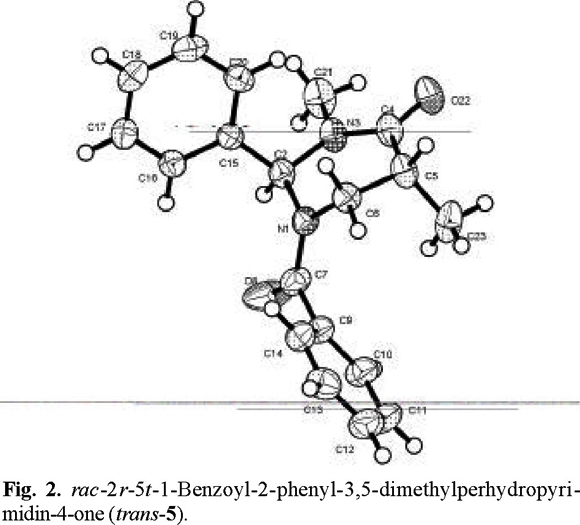
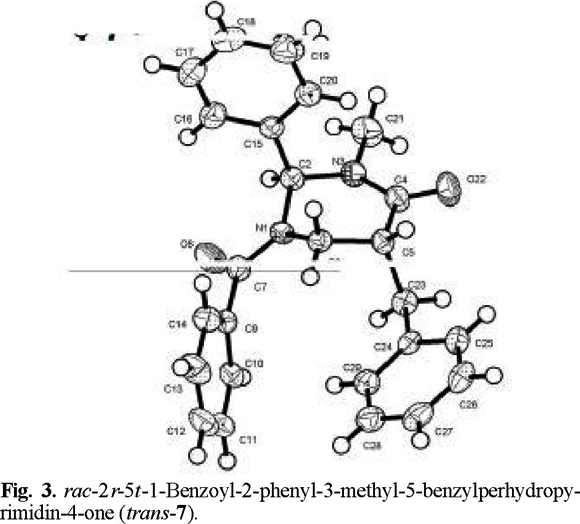
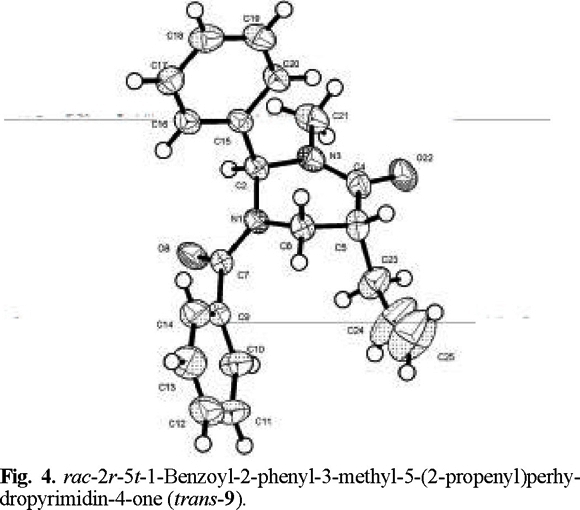
That addition took place preferentially from the side opposite to the phenyl group, to afford the trans products, was determined by X-ray diffraction analysis (Figs. 2-4 (3)).
In addition, we obtained the dimethylated product (6) when the electrophile is CH3I, as consequence of a second addition of CH3I (Eq 1). The solid-state crystal structure of 6 shows that steric repulsion provokes bending of the 2-phenyl ring. (Fig. 5).


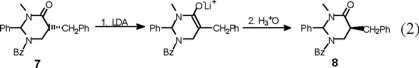
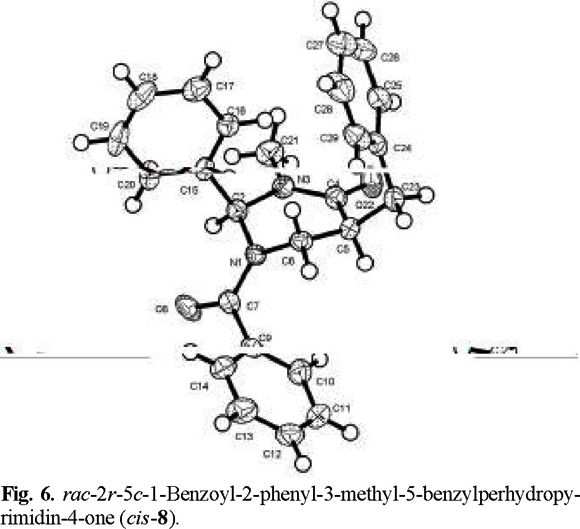
Finally, an X-ray diffraction study of the benzylated derivative (8) (Fig. 6) confirmed its relative configuration as cis, which can be envisaged to result from epimerization at C(5) in adduct (7) (eq 2). Interestingly, the solid-state conformation of cis-8 suggests that the aromatic phenyl rings at C(2) and the benzyl substituent may experience ϖ/ϖ attraction [7].
Finally, no alkylation reaction was observed with isopropyl iodide as electrophile, as consequence of the lower reactivity of the secondary halide.
Conclusions
β-Alanine, an inexpensive and achiral amino acid, was converted efficiently into the racemic perhydropyrimidinone 1. The high diastereoselectivity observed in the addition of enolate 1-Li to electrophiles shows that the chirality center at C(2) supporting the phenyl group induces the stereoselective formation of the new stereogenic center at C(5). These results pave the way to the exploitation of enatiopure analogs of rac-1 in the asymmetric synthesis of α-substituted β-amino acids.
Experimental Section
General. Flasks, stirring bars, and hypodermic needles used for the generation and reactions of organolithiums were dried for 12 h at 120 °C and allowed to cool in a desiccator over anhydrous CaSO4. Anhydrous solvents were obtained by distillation from benzophenone ketyl [8]. The n-BuLi employed was titrated according to the method of Juaristi et al. [9].
TLC: Merck-DC-F254 plates; detection by UV light. Flash column chromatography [10]: Merck silica gel (0.040-0.063 mm).
Melting points: Mel-Temp apparatus; not corrected. 1H NMR spectra: Hitachi: 60 MHz, JEOL: 270 MHz, Varian 200 MHz and 400 MHz. 13C NMR spectra: Varian 50 MHz and 100 MHz. Chemical shifts (δ) are given in parts per million down-field from the internal TMS reference; the coupling constants (J) in hertz.
X-Ray diffraction analysis: CAD4-Enraf-Nonius and APEX-Bruker diffractometer. The structures were solved by direct methods using the program SHELXS [11]. Space groups, cell constants, number of reflections measured, and final R values are collected in Table 2.
Methyl β-Aminopropionate Hydrochloride (2). β-Aminopropionic acid (10 g, 112.3 mmol) in 60 mL of freshly distilled methanol was placed in a round-bottom flask provided with an addition funnel and a magnetic stirrer. The solution was cooled to 0 °C and treated dropwise with 10.64 mL (145.99 mmol) of thionyl chloride. The reaction mixture was stirred at ambient temperature for 1 h, concentrated in a rotary evaporator, and the precipitate was then filtered to afford 15.38 g (98 % yield) of the desired ester as crystals with mp 92-94 °C (lit [12]. mp 94-95 °C). RMN 1H (60 MHz, D2O) δ 2.5 (br, 2H, C(O)-CH2), 3.0 (br, 2H, N-CH2), 3.6 (s, 3H, O-CH3).
N-Methyl β-Aminopropionamide Hydrochloride (3). Methyl β-aminopropionate hydrochloride (15.35 g, 110 mmol) in 70 mL of methanol was placed in a round-bottom flask provided with a magnetic stirrer and an addition funnel. The soluction was cooled to 0 °C and treated dropwise with 16 mL (215 mmol) of aqueous 40 % methylamine. The resulting mixture was stirred at 0 °C for 72 h. The solvent was evaporated to afford 15.08 g (99 % yield) of the desired amide (3) as hygroscopic oil: RMN 1H (60 MHz, D2O,) δ, 2.6 (brs, 2H, C(O)-CH2), 3.0 (s, 3H, N-CH3), 3.1 (br, 2H N-CH2).
β-(N-3-Benzylidenamino)-N-methylpropionamide (4). Amide 3 (15.00 g, 108 mmol) in 70 mL of benzene was placed in a round-bottom flask provided with an addition funnel and a magnetic stirrer. The resulting suspension was treated dropwise and with stirring with 24.1 mL (173.3 mmol) of freshly distilled Et3N, and then with 12.9 mL (127.1 mmol) of benzaldehyde. The reaction mixture was heated to reflux for 17 h, with a Dean-Stark trap being used to collect the water that was generated. The precipitated triethylamine hydrochloride was removed by filtration, and the filtrate was concentrated to afford 20.52 g (~100 % yield) of the crude imine (4) as a brown oil. This product was not purified because is very sensitive to water.
1-Benzoyl-2-phenyl-3-methylperhydropyrimidin-4-one (1). Imine (4) (20.52 g, 108 mmol) in 90 mL of benzene was placed in a round-bottom flask provided with an addition funnel and a magnetic stirrer. The resulting suspension was treated with 12.7 g (104 mmol) of DMAP and with 14.6 mL (127.1 mmol) of benzoyl chloride (dropwise). The reaction mixture was heated to reflux for 3 h. The reaction mixture was allowed to cool to ambient temperature, and the precipitated dimethylaminopyridine hydrochloride was removed by filtration, and the filtrate was concentrated vacuum. The product (1) was purified by flash chromatography (n-hexane / ethyl acetate, 90:10 to 50:50) to furnish 16.84 g (53 % yield) of a white solid, mp 145-149 °C. 1H RMN (270 MHz, CDCl3) δ 2.41 (ddd, Jgem = 17.5 Hz, Jgauche = 5.4 Hz, Jgauche = 2.22 Hz, 1H, C(O)-CH2); 2.54 (ddd, Jgem = 17.5 Hz, Janti = 11.13 Hz, Jgauche = 7.67 Hz, 1H, C(O)-CH2), 2.96 (s, 3H, N-CH3), 3.21 (ddd, Jgem = 13.85 Hz, Janti = 11.13, Jgauche = 5.44 Hz, 1H, N-CH2), 3.75 (brs, 1H, N-CH2), 6.9 (brs, 1H, Ph-CH), 7.3 (m, 10H, Ph y Bz); 13C RMN (50 MHz, CDCl3) δ 31.9 (CH2CO), 33.5 (CH3N), 39.0 (CH2N), 69.0 (PhCH), 126.6 (Cp phenyl), 129.1 (Co phenyl), 129.18 (Cm phenyl), 136.9 (Cipso phenyl), 126.8 (Cm benzoyl), 128.8 (Co benzoyl),130.5 (Cp benzoyl), 134.50 (Cipso benzoyl), 167.7 (Ph-CO),169.9 (NCO).
General Procedure for the Reaction of Pyrimidinone Enolate (1-Li) with Electrophiles. A solution of (i-Pr)2NH (154 mg 1.1 mmol) in 50 mL of THF was cooled down to −78 °C (dry ice / acetone bath) before the slow addition of 0.45 mL (1.1 mmol) of n-BuLi in hexane (2.5 M). The resulting solution was stirred at −78 °C for 20 min ant then treated with 294 mg (1 mmol) of pyrimidinone (1) in 20 mL of THF. The yellow solution formed was stirred at −78 °C for 1 h before the addition of the electrophile (1.2 mmol), and the reaction mixture was stirred at this temperature for 1 h and at ambient temperature for 15 min. Then the mixture was treated with 10 mL of saturated aqueous ammonium chloride. The aqueous phase was separated and extracted three times with 50-mL portions of CH2Cl2. The combined extracts were dried (Na2 SO4), filtered, and evaporated to give the crude product.
rac-2r-5t-1-Benzoyl-2-phenyl-3,5-dimethylperhydropyrimidin-4-one (trans-5) and rac-1-Benzoyl-2-phenyl-3,5,5´-trimethylperhydropyrimidin-4-one (6). The general procedure was followed for the alkylation of 0.588 g (2 mmol) of 1 with 0.13 mL (2.2 mmol) of CH3I. Purification of the crude product by flash chromatography (n-hexane / ethyl acetate, 90:10 to 40:60) afforded 450 mg (76 % yield) of trans-5 and 110 mg (24 % yield) of 6.
cis-5: mp 111-113 °C; 1H RMN (200 MHz, CDCl3) δ 1.18 (br, 3H, CH-CH3), 2.51 (br, 1H, CH-CH3), 3.05 (s, 3H, N-CH3), 3.48 (br, 2H, CH-CH2), 7.4 (br, 1H, Ph-CH), 7.6 (m, 10H, Ph and Bz); 13C RMN (50 MHz, CDCl3) δ 17.16 (CH-CH3), 34.0 (CH-CH3), 36.39 (N-CH3), 46.29 (CH-CH2), 68.93 (Ph-C), 126.3 (Cp phenyl), 129.1 (Co phenyl), 129.3 (Cm phenyl), 137.5 (Cipso phenyl), 127.1 (Cm benzoyl), 128.9 (Co benzoyl), 130.4 (Cp benzoyl), 134.8 (Cipso benzoyl), 170.71 (Ph-CO), 171.98 (N-CO).
6: mp 136-137 °C; 1H RMN (200 MHz, CDCl3) δ 1.12 (br, 6H, C-(CH3)2), 2.95 (s, 3H, N-CH3), 3.29 (br, 2H, N-CH2), 7.1 (br, 1H, Ph-CH), 7.4 (m, 10H, Ph y Bz); 13C RMN (50 MHz, CDCl3) δ 23.5 (C-(CH3)2), 33.6 (N-CH3), 40.1 (C-(CH3)2), 51.4 (N-CH2), 69.2 (Ph-C), 126.6 (Cp phenyl), 129.2 (C o phenyl), 129.3 (Cm phenyl), 137.5 (Cipso phenyl), 127.3 (Cm benzoyl), 128.8 (C o benzoyl), 130.5 (C p benzoyl), 134.6 (Cipso benzoyl), 170.7 (Ph-CO), 174.55 (NCO).
rac-2r-5t-1-Benzoyl-2-phenyl-3-methyl-5-benzylperhydropyrimidin-4-one (trans-7) and rac-2r-5c-1-benzoyl-2-phenyl-3-methyl-5-benzylperhydropyrimidín-4-one (cis-8). The general procedure was followed for the alkylation of 0.588 g (2 mmol) of 1 with 0.24 mL (2.1 mmol) of C6H5 CH2Br. Purification of the crude product by flash chromatography (n-hexane / ethyl acetate, 90:10 to 30:70) afforded 299 mg (40 % yield) of trans-7 and cis-8. The separation of the products was made with a spatula under a microscope.
trans-7: mp 165-166 °C; 1H RMN (250 MHz, CDCl3) δ 2.4 (brs, 1H, CH-CH2Ph), 2.6 (brs, 1H, CH-CH2Ph), 2.7 (brs, 1H, C(O)-CH), 3.1 (brs , 3H, N-CH3), 3.27 (brs, 1H, N-CH2), 3.6 (brs, 1H, N-CH2), 6.5 (brs, 1H, Ph-CH), 7.0-7.6 (m, 15H, 2Ph and Bz ); 13C RMN (100 MHz, CDCl3) δ 34.0 (N-CH3), 36.5 [C(5)], 42.0 [C(6)], 44.5 (CH-CH2Ph), 69.5 [C(2)], 126.5 (Cp phenyl), 127.7 (Co phenyl), 129.1 (Cm phenyl), 137.3 (Cipso phenyl), 129.0 (Co benzoyl), 128.5 (Cm benzoyl), 130.7 (Cp benzoyl), 134.5 (C ipso benzoyl), 128.9 (Co benzyl), 129.3 (Cm benzyl), 126.0 (Cp benzyl), 138.7 (Cipso benzyl), 170.1 (COPh), 170.6 (NCO).
cis-8: mp 243-244 °C; 1H RMN (400 MHz, CDCl3) δ 2.4 (brs, 1H, CH-CH2Ph), 2.6 (brs, 1H, CH-CH2Ph), 2.7 (brs, 1H, C(O)-CH), 3.1 (brs, 3H, N-CH3), 3.27 (brs, 1H, N-CH2), 3.6 (brs, 1H, N-CH2), 6.5 (brs, 1H, Ph-CH), 7.0-7.6 (m, 15H, 2Ph and Bz ); 13C RMN (100 MHz, CDCl3) δ 34.8 (N-CH3), 37.1 [C(5)], 43.2 [C(6)], 44.1 (CH-CH2Ph), 69.3 [C(2)], 126.8 (Cp phenyl), 128.7 (Co phenyl), 129.7 (Cm phenyl), 137.8 (Cipso phenyl), 129.7 (Co benzoyl), 129.2 (Cm benzoyl), 130.9 (Cp benzoyl), 134.5 (C ipso benzoyl), 129.3 (Co benzyl), 130.5 (Cm benzyl), 126.5 (Cp benzyl),138.7 (Cipso benzyl), 169.9 (COPh), 171.6 (NCO).
rac-2r-5t-1-Benzoyl-2-phenyl-3-methyl-5-(2-propenyl)perhydropyrimidin-4-one (trans-9). The general procedure was followed for the alkylation of 0.588 g (2 mmol) of (1) with 0.19 mL (2.1 mmol) of allyl bromide. Purification of the crude product by flash chromatography (n-hexane / ethyl acetate, 90:10 to 40:60) afforded 0.661 g (~100 % yield) of trans-9: mp 100-101 °C; 1H RMN (200 MHz, CDCl3) δ 2.1 (brs, 1H, C1´), 2.4 (brs, 1H, C1´), 2.5 (brs, 1H, C5), 3.0 (s, 3H, N-CH3), 3.4 (brs, 1H, C6), 3.7 (brs, 1H, C6), 4.1 (brs, 1H, C3´), 4.6 (brs, 1H, C4´), 5.4 (brs, 1H, C4´), 7.1 (brs, 1H, Ph-CH), 7.4 (m, 10H, Ph and Bz); 13C RMN (50 MHz, CDCl3) δ 33.8 (CH-C H2- HC=), 35.4 (CH-CH2-HC=), 41.5 (CH3-N), 42.4 (CH2-N), 44.0 [C(6)], 68.9 (CH-Ph), 117.94 (C=CH2), 137.29 (C=CH2), 126.6 (Cp phenyl), 129.2 (Co phenyl), 129.7 (Cm phenyl), 137.5 (Cipso phenyl), 127.3 (Cm benzoyl), 128.8 (Co benzoyl),130.5 (Cp benzoyl), 134.6 (Cipso benzoyl), 170.1 (N-CO), 169.6 (Ph-CO).
Acknowledgment
We are grateful to CONACyT for a scholarship to A.L.H. and for financial support via grant 3559P-E9607.
References
1. Juaristi, E. (Editor) in: Enantioselective Synthesis of β-Amino Acids. Wiley-VCH, New York, 1997, and references cited. [ Links ]
2. (a) Spatola, A. F. in: Chemistry and Biochemistry of Amino Acids, Peptides and Proteins, Ed. (Marcel Dekker, N.Y., 1983) vol. VII, p. 267; [ Links ] (b) R. A. Wiley, D. H. Rich, Med. Res. Rev. 1993, 13, 328. [ Links ]
3. Brunner, J. Chem. Soc. Review 1993, 22, 183-189. [ Links ]
4. (a) Juaristi, E; Quintana, D.; Balderas, M.; García-López, E. Tetrahedron: Asymmetry 1996, 7, 2233-2246. [ Links ] (b) Juaristi, E.; López-Ruiz, H.; Madrigal, D.; Ramírez-Quirós, Y.; Escalante, J. J. Org. Chem. 1998, 63, 4706-4710. [ Links ] (c) Juaristi, E. Enclyclopedia of Reagents for Organic Synthesis, Paquette, L.A. (Editor), Wiley New York, in press. [ Links ] (d) Ramírez-Quirós, Y.; Balderas, M.; Escalante, J.; Quintana, D.; Gallardo, I.; Madrigal, D.; Molins, E. and Juaristi, E. J. Org. Chem., 1999, 64, 8668-8680. [ Links ]
5. Cardillo, G. and Tomasini, C. in: Enantioselective Synthesis of β-Amino Acids, Juaristi, E. (Editor), Wiley-VCH, New York, 1997, 211-248. [ Links ]
6. Konopelski, J. in: Enantioselective Synthesis of β-Amino Acids, Juaristi, E. (Editor), Wiley-VCH, New York, 1997, 249-260. [ Links ]
7. See, for example: Jorgensen, W.L.; Severance, D.L. J. Am. Chem. Soc. 1990, 112, 4768-4774. [ Links ]
8. Brown, H.C. in: Organic Synthesis via Boranes; Wiley: New York, 1975, 256. [ Links ]
9. Juaristi, E.; Martínez-Richa, A.; García-Rivera, A.; Cruz-Sánchez, J.S. J. Org. Chem. 1983, 48, 2603-2606. [ Links ]
10. Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923-2925. [ Links ]
11. Sheldrick , G. M. SHELX97, Programs for Crystal Structure Analysis, release 97-2; Institute for Inorganic Chemistry der Universität: Tommanstrasse 4, D-3400 Göttingen, Germany, 1998. [ Links ]
12. Dictionary of Organic Compounds; Eyre & Spottiswoode Publishers: London, 1965, Vol. 1, p. 206. [ Links ]

