










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.45 no.4 Ciudad de México oct./dic. 2001
Investigación
La paradoja de Gibbs
Leopoldo García-Colín Scherer*
Departamento de Física, Universidad Autónoma Metropolitana-Iztapalapa, México 09340, D. F.
Recibido el 22 de mayo del 2001.
Aceptado el 19 de junio del 2001.
Este artículo está dedicado a mi gran amigo y colega Fernando Walls Armijo con motivo de su septuagésimo aniversario.
Resumen
Se realiza una reflexión referente a las sutilezas de la Termodinámica, en particular, sobre la paradoja de Gibbs. El cálculo del cambio de entropía en el proceso de difusión isobárico e isotérmico de dos gases ideales diferentes permite suponer que cuando el proceso se lleva a cabo con dos muestras del mismo gas, sigue habiendo mezclado, lo cual constituye una paradoja. Se demuestra que tal paradoja en realidad no existe, sino que es el resultado de un pobre entendimiento y de una mala aplicación de las leyes de la Termodinámica.
Palabras clave: Paradoja, Gibbs, difusión, gases ideales, Termodinámica.
Abstract
A reflection on the subtleties of Thermodynamics is made, in particular, on the Gibbs paradox. The calculation of the entropy change of the isobaric and isothermic process of difussion of two ideal gases allows to suppose that when the process takes place with two samples of the same gas, the mixing exists, which constitutes a paradox. It is shown that actually the paradox does not exist, and in fact, it is the result of a poor understanding and a wrong application of the laws of Thermodynamics.
Keywords: Paradox, Gibbs, difussion, ideal gases, Thermodynamics.
Introducción
Mi muy estimado Fernando: Hace más de cincuenta años que nos conocemos y disfrutamos épocas de estudiantes llenas de episodios, casi todos de grata memoria y mucho antes de que nos encauzáramos por el camino de la Ciencia. Uno de ellos, y que seguramente recuerdas tan bien como yo, me dejó muchas enseñanzas que en cierto modo motivan este artículo. En 1949, en aquellos entonces nuevos y flamantes edificios de la calle de Nicolás San Juan (629 o 729, no recuerdo con precisión) cursábamos el segundo año de la carrera, que a esas alturas no distinguía química de ingeniería química, con maestros tan ilustres como don Manuel Rangel, Eustaquio Cepeda, Fermín Athié y Alejandro Purón de la Borbolla. Este último nos dio un curso de Termodinámica del cual yo no sé tú que aprendiste, pero yo, no mucho. Sabía hacer balances de masa, energía, calor y ciertamente a manejar con destreza la regla de cálculo (¡no había computadoras!) y entreveía más o menos el significado de la primera ley de la termodinámica pero nada más. Si alguien al finalizar el curso me hubiera preguntado sobre las leyes de la termodinámica, me hubiera quedado mudo. Al año siguiente, recordarás, el pleito Anfossi-Cepeda nos llevó galantemente a la entonces Escuela Nacional de Ciencias Químicas de la UNAM allá en Tacuba, cerca del árbol de la Noche Triste, donde no se porqué razón tomé el curso de Termodinámica con don Efrén Fierro. Fuera del concepto del infinito que alguna vez ilustró profusamente, tampoco aprendí gran cosa; pero por otra razón, igualmente misteriosa, leí el libro de Termodinámica de Max Planck y ahí empezó mi calvario. Tú te dedicaste a la Química Orgánica y como buen cazador y tirador, le has sido fiel toda la vida, pero yo he ido saltando de mata en mata dentro de esa borrosa frontera entre la Física y la Química, pero sin dejar a la Termodinámica de lado. Llevo entonces cincuenta años estudiando esta materia y te confieso que a veces pienso que no he llegado a entenderla y dominarla completamente. Dentro de su aparente simplicidad y desprovista de herramientas matemáticas sofisticadas y esotéricas, es terriblemente sutil, engañosa ¡y muy soberbia! Sobrevivió a las dos grandes revoluciones científicas de la física contemporánea, la física cuántica y la teoría de la relatividad. Es por ello, y en recuerdo de nuestras viejas memorias, que quiero compartir contigo una experiencia en esta materia que hoy en día, a pesar de su simplicidad, si no es ignorada, es totalmente tergiversada por los muchos autores de libros de texto sobre la materia. La experiencia está basada en lo que los autores de libros de físico-química llaman la paradoja de Gibbs y algunos físicos, la llaman paradoja del N!
La paradoja de Gibbs
Tomemos un sistema muy simple. En dos recipientes cuyos volúmenes son V1 y V2 colocamos N1 moléculas de un gas de cierta especie 1 y N2 moléculas de otro gas de especie 2, respectivamente. Los dos recipientes se restringen a tener la misma temperatura T y presión p. Por facilidad en los cálculos supondremos a los dos gases ideales, Xe y Ar, por ejemplo. Ahora supongamos que ponemos ambos recipientes en contacto entre sí y a través de la pared común abrimos un pequeño orificio dejando que los gases se mezclen entre sí. Esto genera un proceso isotérmico e isobárico, p y T permanecen constantes, la difusión es muy lenta, el proceso es reversible y al final del proceso las N1 + N2 moléculas de ambas especies pueden compartir el volumen total de los dos recipientes. La pregunta es si hay un cambio en la entropía de los gases debido al mezclado y de ser así, cómo calcularla. Para ello voy a proceder como se hace en la gran mayoría (¡si no en todos!) los libros que tocan este problema. Llamando S y U a la entropía y energía interna de un sistema cualquiera, la ecuación fundamental de la termodinámica establece que para todo proceso infinitesimal y reversible,

si las variables independientes son U y V. Esta ecuación tiene cinco variables, dos son independientes y por lo tanto necesitamos dos ecuaciones adicionales para integrarla. Por ser los gases ideales, el experimento nos dice que la presión y la energía interna están relacionadas con la temperatura de acuerdo con las ecuaciones
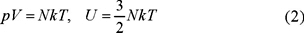
donde k ≡ R / N0 es la constante de Boltzmann, R la constante universal de los gases y N0 el número de Avogadro. Sustituyendo (2) en (1) obtenemos para dS una forma diferencial exacta, como lo exigen las leyes de la termodinámica, cuya integral es
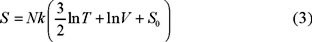
donde S0 es la constante de integración.
Es con este resultado que los expositores de esta materia intentan responder la pregunta planteada. En efecto, la entropía de cada uno de los gases que intervienen en nuestro experimento, cuando ocupan sus sendos recipientes, está dada por,
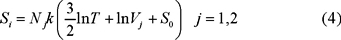
y después del mezclado la entropía corresponde a la de un gas formado por N = N1 + N2 moléculas ocupando un volumen V = V1 + V2 a la misma temperatura. Entonces en el estado final,
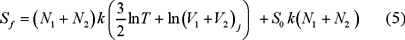
La entropía inicial es Si = S1 + S2 luego el cambio en la entropía se obtiene restando a la ec. (5) la entropía inicial Si para obtener que
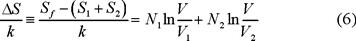
Si en la ec. (6) tomamos N1 = N2 = N0, una mol de cada gas y suponemos que V1 = V2 = V/2 tenemos que

La entropía de mezclado es un número que no depende de nada, ni de la naturaleza de las especies, la forma de los recipientes, etc., cuando el proceso es isotérmico e isobárico. Esto quiere decir que si hacemos una serie de experimentos con un gas formado por átomos A y otros formados por sus respectivos isótopos Ai (i = 1, 2...) de manera que A y Ai son diferentes y el mezclado tenga sentido, la ec. (7) es plausible. Pero si repetimos el experimento con dos muestras del mismo gas ¡según (7) sigue habiendo "mezclado"! Esto es a todas luces absurdo. Además si hiciéramos el experimento con n gases ocupando inicialmente volúmenes V1 = V2 = ... = Vn = V/n y N1 = N2 = ... = N0,

independientemente de si los gases son o no idénticos entre sí. Estos resultados constituyen la llamada "paradoja de Gibbs".
Es un ejercicio ilustrativo e interesante ver en la literatura la enorme cantidad de teorías, pseudo teorías, conceptos estrafalarios sobre distinguibilidad e indistinguibilidad de partículas microscópicas, clásicas y cúanticas, etc., que se han generado en el proceso de entender las ecs. (7) y (8). No entraré en estos detalles aquí por razones de espacio[1]. Lo que quiero mostrar es que la paradoja de Gibbs no existe y es el resultado de una mala aplicación de las leyes de la termodinámica [2].
La solución de la paradoja de Gibbs
En el aprendizaje de la termodinámica de sistemas en equilibrio, o termostática como debería llamársele, es imprescindible digerir el hecho de que la ec. (1) establece que la entropía es un potencial termodinámico en la representación en la cual las variables de estado son extensivas. En el caso de un fluido esto nos dice que S = S(U, V, N). Cuando un proceso ocurre en un sistema cerrado (N = cte.), dN = 0 y por ello el término diferencial en esta variable no aparece explícitamente en dicha ecuación. Ahora bien, también se nos enseña [3] que la dependencia de la entropía con U, ∇ y N no es arbitraria, S debe satisfacer la regla de Euler para funciones homogéneas de primer grado. Esto quiere decir que para todo número real λ > 0

que pocos autores subrayan. Además como la entropía es extensiva, para un conjunto arbitrario de r sistemas en equilibrio,

Las ecs. (9) y (10) son fundamentales para aclarar nuestra paradoja. En efecto, reescribamos la ec. (3) en el lenguaje apropiado usando la ec. (2) para U. Obtenemos que, eliminando la temperatura T,

donde S'0 = S0 − 3/2 ln k es una nueva constante. Es obvio, a todas luces, que la ec. (11) es incorrecta pues no satisface la ec. (9), ln (λV) ≠ λ ln V. ¿Dónde está la falla? Ahora mi querido amigo, nos acordamos de lo que sí nos enseñó y muy bien el venerable Sr. Rangel en las materias de cálculo. En los capítulos sobre funciones de varias variables, continuas, diferenciables y decentes por todos lados, en nuestro viejo texto, el Granville-Smith hay un teorema que dice que si f(x, y, z) es decente y sólo hago con ella operaciones en el plano xy (diferenciar, integrar, etc.) jamás podré determinar su dependencia en z. Si esta propiedad la llevamos a la ec. (1), implica que jamás podré determinar la dependencia de S con N en tanto realice operaciones con sistemas cerrados. En el lenguaje matemático esto implica que la constante S0 es todavía una función indeterminada de N ¿Y cómo la determino? Pues uso como condición a la frontera que dicha constante me genere una forma para S que sea consistente con la ec. (9). No se necesita pensar mucho para ver que la única opción es tomar

donde c es un número arbitrario que no depende de V, de U ni de N. Introduciendo (12) en (11) encuentro que
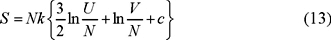
que obviamente satisface la propiedad de homogeneidad. Regresando a la forma de la ec. (3) tenemos que
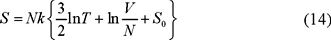
donde S0 es estrictamente un número.
La ec. (14) es más convincente para repetir el cálculo del cambio de entropía ya que éste se lleva a cabo isotérmica e isobáricamente. Ahora repetimos la operación de calcular ΔS = Sf − Si en dos casos diferentes.
a) Gases idénticos
La entropía no cambia, antes del mezclado está dada por S1 + S2 donde
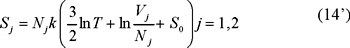
según la ec. (14). De acuerdo con esta misma ecuación, la entropía final es la de un gas con N = N1 + N2 moléculas de la misma especie ocupando el volumen V = V1 + V2 de manera que
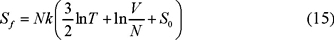
de modo que, usando las ecs. (14') y (15) obtenemos,

pero 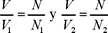 por la ec. (1) y por lo tanto,
por la ec. (1) y por lo tanto,
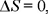
como debería ser; al ser los gases idénticos no hay mezclado y por tanto no hay cambio en la entropía.
b) Gases diferentes
En este caso la entropía inicial vuelve a ser la misma, pero la final no; para calcularla usamos la ec. (10), la entropía final es la suma de las entropías de dos gases ideales: uno con N1 moléculas y el otro con N2 moléculas ocupando un volumen V. Esto es
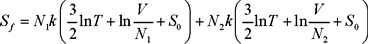
de manera que
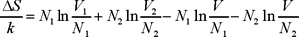
expresión que obviamente se reduce a
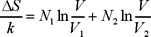
que es la ec. (6). Cuando los gases son diferentes tiene sentido hablar de mezclado y en el caso particular discutido en la sección anterior, ΔS se reduce a un número.
El lector escéptico todavía podría pensar que esto es una coincidencia afortunada por usar la forma (14) para la entropía, esto es S(T, V, N). No es así. Si se usa la ec. (1) para eliminar V/N = kT / p de dicha expresión se obtiene que

que es la fórmula estándar para S en la representación T, p y N. El lector puede repetir todos los cálculos [4] y convencerse por sí mismo que los resultados son los mismos. Igualmente ocurre si se toma S = S(p, V, N). El resultado, cual debe ser, no depende de la representación elegida para describir los estados del gas.
Como un comentario adicional, la ecuación (12) exhibe la razón por la cual los físicos llaman a esta paradoja la del N!.
En efecto, haciendo c = 1, arbitrariamente,
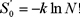
que por la aproximación de Stirling es la ec. (12), como N es muy grande está perfectamente justificada. Ahora bien, en termodinámica estadística, cuando se usa el método de Boltzmann para relacionar la entropía con la distribución más probable de las moléculas en sus microestados aparece un N! [5]. El cálculo conduce a la forma errónea de la entropía dada por la ec. (3). Para obtener la forma correcta, ec. (14) hay que dividir entre este factor arbitrariamente. Como su aparición proviene de "distinguir" como N objetos (moléculas) pueden acomodarse en diferentes niveles energéticos, su eliminación se asocia la concepto de "indistinguibilidad". Esto es completamente erróneo. Uno divide por N! para llegar a una forma correcta para la entropía según la ec. (9) y nada más. Esta operación es proporcionada por la naturaleza misma al reconocer que, según la mecánica cuántica, en la naturaleza sólo existen fermiones y bosones. Con ello se puede demostrar (ver ref [5]) que N! estados "clásicos" corresponden a un solo estado cuántico y con ello aparece la forma correcta para la entropía. Esto nada tiene que ver con la distinguibilidad o indistinguibilidad de partículas clásicas o cuánticas.
Concluyendo, la paradoja de Gibbs no existe, sólo aparece debido a un pobre entendimiento de las leyes de la termostática. Son precisamente estos detalles que hacen a esta materia un tanto sutil pero de una belleza y generalidad incomparables.
Bibliografía
1. Es imposible dar una lista exhaustiva de las obras que de una manera u otra tocan este tema. Un muestreo representativo es el siguiente:
Zemansky, M. W. Heat and Thermodynamics; McGraw Hill Book Co., 5ª ed., N. Y., 1968, 561-563. [ Links ]
Bridgman, P. W. The Nature of Thermodynamics; Peter Smith, Gluocester, Mass, 1969, 166-170. [ Links ]
Fermi, E. Thermodynamics; Dover Publications, N. Y., 1957, Sec. 32, 147. [ Links ]
Kestin, J. A Course in Thermodynamics, Vol. I; Mc Graw Hill Book Co., N. Y., 1979, 578. [ Links ]
Reichl, L. A Modern Course in Statistical Physics; University of Texas Press, Austin, 1980, 68. [ Links ]
Pesic, P. D. Am. J. Phys. 1991, 59, 971, 975. [ Links ]
2. van Kampen, N. G. en Essays in Theoretical Physics (in Honour of D. ter Haar) Pergamon Press, Oxford, 1984, 303. [ Links ]
3. García-Colín S., L. Introducción a la termodinámica clásica; Ed. Trillas, 4ª ed., México D.F., 1996, cap. 8. [ Links ]
4. García-Colín S., L. Introducción a la termodinámica de sistemas abiertos; El Colegio Nacional, 2ª ed. 2001 (en prensa). [ Links ]
5. García-Colín S., L. Termodinámica estadística. UAM-Iztapalapa, México D.F., 1995, Cap. 2. [ Links ] En esta referencia el tratamiento de la paradoja de Gibbs es incorrecto.
Nota
*Miembro del Colegio Nacional.

