










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.45 no.1 Ciudad de México ene./mar. 2001
Investigación
Asymmetric Synthesis of Naturally Occurring β-Hydroxyamides (R)-Tembamide and (R)-Aegeline
Gerardo Aguirre, Araceli Salgado-Rodríguez, Lucía Z. Flores-López, Miguel Parra-Hake y Ratnasamy Somanathan*
Centro de Graduados e Investigación del Instituto Tecnológico de Tijuana, Blvd. Industrial S/N, Mesa de Otay. Tijuana 2200, Baja California Norte, México. Apdo. Postal 1166, 22,000. Tel: (66) 233-772; Fax: (66) 234-043. E-mail: mparra@tectijuana.mx
Recibido el 17 de febrero del 2001.
Aceptado el 16 de marzo del 2001.
Abstract
Chiral cyanohydrins were synthesized from anisaldehyde and trimethylsilylcyanide catalyzed by a chiral Schiff-base titanium complex. Cyanohydrins were converted into chiral the ß-hydroxyamides, (R)-Tembamide and (R)-Aegeline.
Keywords: Chiral cyanohydrins, chiral Schiff base-titanium complex, ß-hydroxyamides.
Resumen
Se sintetizaron cianohidrinas quirales a partir de anisaldehído y cianuro de trimetilsililo por medio de la reacción catalizada con un complejo de titanio y una base de Schiff quiral. Las cianohidrinas fueron convertidas a las ß-hydroxyamidas quirales, (R)-Tembamida y (R)-Aegelina.
Palabras clave: Cianohidrinas quirales, bases de Schiff quirales-complejo de titanio, ß-hydroxyamidas.
Introduction
(R)-(−)-Tembamide (1) and (R)-(−)-aegeline (2) are two naturally occurring ß-hydroxyamides isolated from Fagara hyemalis (St. Hill) Engler and Aegele marmelos Correa, respectively, belonging to the family Rutaceae [1-4]. These β-hydroxyamides have been reported to have insecticide and adrenaline-like activity. Extracts of Aegle marmelos, containing tembamide (1), have been used in the Indian traditional medicine as a control for hypoglycemia [5]. One report also claims that the leaves of Aegle marmelos are used in Bangladesh for fertility control [6].

In addition to their varied biological properties, these ß-hydroxyamides are possible intermediates in the plant biosynthesis of enamides (3) and oxazoles (4), as they have been isolated from several species of the same family [7-10]. The enamides and oxazoles may be formed from the ß-hydroxyamide (1 and 2) by a simple dehydration process to enamide (3) or by an internal SN2 type displacement to oxazoline followed by oxidation to oxazole (4) (Scheme 1). In a recent publication we have shown the facile formation of oxazolines from chiral ß-hydroxyamides (erythro- and threo-) by chemical or thermal induced cyclization processes, where the chirality in the oxazoline is either retained or inverted [11]. To our knowledge, the biosynthetic interconnection between these compounds has not been fully established in the family Rutaceae.
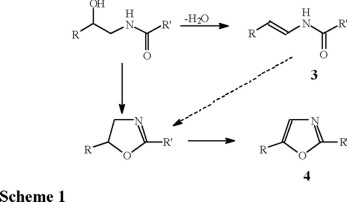
These two β-hydroxyamides (1 and 2) were isolated from Nature as racemic mixtures and later resolved into their enantiomers as tartrate salts [12]. A previous report from this laboratory described the synthesis of racemic tembamide (1) and aegeline (2) from the cyanohydrin obtained by the addition of trimethylsilylcyanide to anisaldehyde with catalysis by zinc iodide [13]. More recent advances now allow addition of trimethylsilylcyanide to carbonyl groups in an enantioselective fashion, using chiral Lewis acid catalysts [14-17]. In view of their interesting biological and possible biosynthetic roles, we embarked on the enantioselective synthesis of tembamide (1) and aegeline (2). Here we report the synthesis of a chiral cyanohydrin and its transformation to the chiral ß-hydroxyamides, tembamide (1) and aegeline (2).
Results and discussion
Recently we reported the asymmetric addition of trimethylsilylcyanide to benzaldehyde catalyzed by titanium (IV)-Schiff base complexes derived from a chiral cis-indanol system [17]. In this study we discovered that ligands 5 and 6 gave the best enantioselectivity in the hydrocyanation reaction. We believe the reason for this high enantioselectivity is due to the rigid five-membered ring backbone of the cis-indanol and the presence of two chiral centers, probably enhancing the chirality in the final cyanohydrin. Further, the X-ray structure of ligand 5 [18] (Fig. 1) suggests that the indanol ring may sterically hinder one face of the carbonyl from cyanide ion attack in the transition state involving the titanium complex.


Having shown the versatility of these chiral Schiff base ligands, we turned our attention to exploiting these ligands in the asymmetric synthesis of a variety of oxygenated natural products, such as ß-hydroxyamides 1 and 2. Here we report the use of ligands 5 and 6 and titanium tetraisopropoxide as catalyst in the addition of trimethylsilylcyanide to anisaldehyde (7), giving the cyanohydrin in 95 and 89 % ee, respectively. Reduction of the cyanohydrin (8) with diborane gave the ß-amino alcohol (9) and subsequent acylation with benzoyl and cinnamoyl chlorides gave tembamide (1) and aegeline (2), respectively, in high optical purity (95 % ee) (Scheme 2). Amides (1) and (2) were characterized by 1H, 13C and X-ray (Figs. 2 and 3) [19, 20].



Conclusions
Chiral cyanohydrins are versatile synthetic intermediates which can be converted to α-hydroxy carboxylic acids, α-hydroxy aldehydes, α-hydroxy ketones. Here we have demonstrated the enantioselective synthesis of a cyanohydrin derived from anisaldehyde and its transformation to the chiral ß-aminoalcohol (9) and ß-hydroxyamides 1 and 2 in high optical purity. Although our method is complementary to the one reported by Jackson and co-workers employing a chiral dipeptide and hydrogen cyanide [21], our method has several advantages: it is not necessary to use HCN; in our case using trimethylsilylcyanide as the source for cyanide makes it easier to handle, and the trimethylsilyloxycyanohydrin adduct (8a) itself can be purified by distillation under reduced pressure. Further, cyanohydrin with the S-configuration can also be synthesized conveniently using ligand 5 with the S,R-configuration.
Experimental
1H and 13C NMR spectra were recorded on a Varian Gemini 200 Spectrometer, and on a Varian Unity Inova 500 MHz spectrometer with TMS as internal standard. X-ray data were collected on a Siemens P4 difractometer. Structure solution were performed by direct methods, and structure refinement was done with the program SHELXS [22]. IR spectra were obtained on a Perkin-Elmer 1600 series spectrometer. Enantiomeric excesses were determined using a Hewlet-Packard 6890 gas chromatograph with a 30 m Supelco ß-DEX column. The optical rotations were obtained on a Rudolph Research Flanders automatic polarimeter. Unless otherwise specified all reagents were purchased from Aldrich Chemical Co. and used without further purification.
(R)-(−)-2(4-Methoxyphenyl)-2-(trimethylsilyloxy)-acetonitrile, (8a). Under a nitrogen atmosphere, ligand 5 [17] (0.264 g, 0.85 mmol) was stirred with 6 mL CH2Cl2 at 23 °C. To the stirred solution Ti(O-i-Pr)4 (0.244 g, 0.85 mmol) was added and the mixture stirred at room temperature for 1 h. The solution was then cooled to −780 °C and trimethylsilylcyanide (0.72 mL, 5.65 mmol) and anisaldehyde (0.56 mL, 4.6 mmol) were added and the mixture stirred at −78 °C for 36 h. The crude mixture was passed through a short column of silica gel and the product concentrated and subjected to short path distillation using Kugelrohr oven, fraction boiling at 95 °C/3 mm Hg was collected. Orange-yellow liquid (0.82 g 77 % yield); bp 95 °C/3 mm Hg; [α]D = + 21.80° (c = 1.00, CHCl3) {lit. [21], [α]D = +22° (c = 1.00, CHCl3)}; IR (film): 1600, 1512,1460, 1250 cm−1; 1H NMR (200 MHz, CDCl3): δ 7.37 (d, 2H, J = 7.9 Hz), 6.90 (d, 2H, J = 7.9Hz), 5.50 (s, 1H), 3.77 (s, 3H) and 0.18 (s, 9H); 13C NMR (50 MHz, CDCl3): δ 160.28, 128.37, 127.86, 119.30, 114.17, 63.17, 55.14 and 0.42.
(R)-(+)-2-Hydroxy-2-(4-methoxyphenyl) acetonitrile, (8b). Compound 8a (0.50 g) was stirred with 1M HCl (10 mL) for 4 h, and the product extracted into dichloromethane. The crude material was recrystallized from dichloromethane/hexane to give 8b (0.48 g) in 95 % yield and 95 % ee. Enantiomeric excess was determined by derivatizing the alcohol with acetic anhydride and injecting into a Hewlet-Packard 6890 gas chromatograph with a 30 m Supelco β-DEX column. The retention times of the enantiomers were 15.17 min (R) and 15.45 min (S). Mp: 84-87 °C (lit. [23], 74-76 °C); [α]D = + 47.50° (c = 1, CHCl3), {lit. [23], [α]D = + 48.80° (c = 1, CHCl3)}. IR (KBr): 3398, 2247 cm−1; 1H NMR (200 MHz, CDCl3): δ 7.42 (d, 2H, J = 8.9Hz), 6.93 (d, 2H, J = 8.9Hz), 5.45 (s, 1H), 3.81 (s, 3H) and 3.21 (brs, 1H); 13C (50 MHz, CDCl3): δ 160.78, 128.32, 127.66, 119.40, 114.57, 63.22 and 55.35.
Reduction of (8b). Compound 8b (0.5 g, 3 mmol) in dry ether (50 mL) was added to a stirred ice-cold solution of BH3:S Me2 solution (3.07 mL, 6 mmol). The mixture was allowed to stand at room temperature overnight and the excess BH3 was destroyed by addition of methanol. Solvent was removed under reduced pressure to give a brownish oil in 74 % yield (0.38 g). 1H NMR (200 MHz, CDCl3): δ 7.13 (d, 2H, J = 7.9 Hz), 6.76 (d, 2H, J = 7.9 Hz), 4.42 (m, 1H), 3.71 (s, 3H), 3.1 (brs. 1H) and 2.62 (m, 2H); 13C (50 MHz, CDCl3): δ 158.53, 134.89, 126.77, 113.32, 73.49, 54.74 and 48.86.
Acylation of amino alcohol (9). Without further purification, the crude amino alcohol 9 was reacted with benzoyl chloride and trans-cinnamoyl chloride under Schotten-Bauman conditions [13] to give (R)-(−)-tembamide (1) and (R)-(−)-aegeline (2) [13] in 83 % and 85 % yields, respectively.
(R)-(−)-Tembamide (1). White solid (0.53 g, 83 %), Mp: 144-146 °C (lit [12], 156-157 °C); [α]D = −58.40° (c = 0.53, CHCl3), {lit. [12], [α]D = −55.31° (c = 0.5, CHCl3)}. IR(KBr): 3345, 3299, 1633, 1245 and 1031 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.74 (d, 2H, J = 7.1 Hz), 7.48 (t, 1 H, J = 7.4 Hz), 7.39 (t, 2H, J = 7.8 Hz), 7.29 (d, 2H, J = 8.5 Hz), 6.88 (d, 2H, J = 8.7 Hz), 6.69 (br. s 1H), 4.85 (dd, 1H, J1 = 3.2 Hz and J2 = 7.8 Hz) 3.88-3.81 (m, 1H), 3.77 (s, 3H), 3.53-3.39 (m, 1H); 13C (125 MHz, CDCl3): δ 168.52, 159.31, 134.12, 133.89, 131.61, 128.55, 127.07, 113.43, 73.19, 55.27 and 47.74.
(R)-(−)-Aegeline (2). White solid (0.58 g, 85 %); Mp: 193-195 °C (lit. [12], 196-197 °C); [α]D = −39.30° (c = 0.45, CHCl3), {lit. [12], [α]D = −35.1° (c = 0.4, CHCl3)}. IR(KBr): 3368, 3283, 1653, 1596, 1241, 1074 and 1032 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.57 (d, 1H, J = 15.62 Hz), 7.50 (d, 2H, J = 8 Hz), 7.43 (br. t, 1H, J = Hz), 7.36-7.35 (m, 3H), 7.33 (d, 2H, J = 8 Hz), 6.77 (d, 2H, J = 8 Hz), 6.56 (d, 1H, J = 15.62 Hz), 5.08 (br. s, 1H), 4.79 (dd, 1H, J1 = 3.39 Hz and J2 = 8.19 Hz), 3.80 (s, 3H), 3.72 (ddd, 1H, J1 = 3.5 Hz, J2 = 6.9 Hz and J3 = 13.79 Hz) and 3.35 (ddd, 1H, J1 = 4.6 Hz, J2 = 8.5 Hz and J3 = 13.8 Hz); 13C NMR (125 MHz, CDCl3): δ 165.67, 138.89, 134.15, 127.88, 127.04, 126.73, 126.28, 120.71, 112.65, 71.21, 54.18 and 46.74.
Supporting Information Available. The X-ray crystal structure data for 1, 2 and 5 are available. Tables of final atomic coordinates for the non-hydrogen atoms, anisotropic thermal parameters, complete list of bond distances and angles and complete crystallographic data are included [24].
Acknowledgments
We gratefully acknowledge support for this project by COSNET grant (735.99P) 1998, CONACyT Proyecto Infraestructura (F264-E9207) for funding of an X-ray diffractometer for Instituto Tecnológico de Tijuana and also for fellowship by CONACyT for A.S.R. and L.Z.F.L.
References
1. Kuck, A. M.; Albonico, S. M.; Deulofeu, V. Chem. and Ind. 1966, 945-946. [ Links ]
2. Chatterjee, A.; Bose, S. J. Indian. Chem. Soc. 1952, 29, 425. [ Links ]
3. Johns, S. R.; Lamberton, J. A.; Price, J. R. Aust. J. Chem. 1967, 20, 2795-2797. [ Links ]
4. Johns, S. R.; Lamberton, J. R; Tweeddale, H. J.; Willing, R. I. Aust. J . Chem. 1969, 22, 2233-2236. [ Links ]
5. Shoeb, A.; Kapil, R. S.; Popli, S. P. Phytochemistry 1973, 12, 2071-2072. [ Links ]
6. Govindachari, T. R.; Premila, M. S. Phytochemistry 1983, 22, 755-757. [ Links ]
7. Brossi A., The Alkaloids, Chemistry and Pharmacology. Vol. 35, Edit. Academic Press, 1989. [ Links ]
8. Chatterjee, A.; Chakrabarty, M.; Kundu, A. B. Aust. J. Chem. 1975, 28, 457. [ Links ]
9. Burke, B. A.; Perkins, H. Tetrahedron Lett. 1978, 2723. [ Links ]
10. Burke, B. A.; Philip, S. Heterocycles 1985, 23, 257-260. [ Links ]
11. Somanathan, R.; Aguilar, H. R.; Rivero, I. A.; Aguirre, G.; Hellberg, L. H.; Yu, Z.; Thomas A. J .Chem. Res. (s) (in print).
12. Albonico, S. M.; Kuck, A. M.; Deulofeu, V. J. Chem. Soc. (C). 1967, 1327-1328. [ Links ]
13. Somanathan, R.; Aguilar, H. R.; Ventura, G. R. Smith, K. M. Synth. Commun. 1983, 13, 273-280. [ Links ]
14. North, M. In Comprehensive Organic Functional Group Transformations; Eds. Katritzky, A.R.; Meth-Cohn, O.; Rees, C. W.; Pattenden, G. Pergmon Press: Oxford. 1995, Vol. 3, Chapter 18. [ Links ]
15. North, M. Synlett 1993, 807-820. [ Links ]
16. Effenberger, F. Angew. Chem. Int. Ed. Engl. 1994, 33, 1555-1564. [ Links ]
17. Flores-Lopez, L. Z.; Parra-Hake, M.; Somanathan, R.; Walsh, P. J. Organometallics 2000, 19, 2153-2160. [ Links ]
18. X-ray quality crystals were obtained from a methanol-hexane solution of (5). X-ray analysis: empirical formula C20H23NO2, F. W. 309.39, T = 294 K, orthorhombic, space group P212121, a = 9.0434(12) Å, b = 10.259(2) Å, c = 18.788(3) Å, α = 90°, ß = 90°, γ = 90°, V = 1743.0(5) Å3, z = 4, Dc = 1.179 mg / m3, F (000) = 664, λ = 0.71073 Å, µ= 0.075 mm−1, 2.17° < 2ϑ < 30.00, R1 = 0.1079, wR2 = 0.2006, largest diff. Peak and hole 0.200 and −0.196 eÅ−3.
19. X-ray quality crystals were obtained by slow evaporation of a CDCl 3 solution of (1). X-ray analysis: empirical formula C16H17NO3, F.W. 271.31, T = 294 K, monoclinic, space group I2 / a, a = 19.188(14) Å, b = 9.560(4) Å, c = 30.90(3) Å, α = 90°, ß = 95.45(4)°, γ = 90°, V = 5643(7) Å3, z = 16, Dc = 1.277 mg / m3, F (000) = 2304, λ = 0.71073 Å, µ = 0.088 mm−1, 2.13° < 2ϑ < 22.61, R1 = 0.0968, wR2 = 0.2285, largest diff. Peak and hole 0.315 and −0.357 eÅ−3.
20. X-ray quality crystals were obtained by slow evaporation of a CDCl 3 solution of (2). X-ray analysis: empirical formula C18H19NO3, F.W. 297.34, T = 293 K, monoclinic, space group P21, a = 6.866(5) Å, b = 8.944(4) Å, c = 12.942(12) Å, α = 90°, ß = 90.21(4)°, γ = 90°, V = 794.7(10) Å3, z = 2, Dc = 1.243 mg / m3, F (000)= 316, λ = 0.71073 Å, µ= 0.085 mm−1, 1.57 ° < 2ϑ < 22.49, R1 = 0.0851, wR2 = 0.1729, largest diff. Peak and hole 0.199 and −0.205 eÅ−3.
21. Jackson, W. R.; Jacobs, H. A Jayatileke, G. S.; Mathews, B. R.; Watson, K. G. Aust. J. Chem. 1990, 43, 2045-2062. [ Links ]
22. Sheldrick, G. M. SHELXTL PC Version 5.03. An Integrated System for Solving, Refining and Displaying Crystal Structures from Diffractometer Data; Siemens Analytical X-ray Instruments, Inc.: Madison, WI, 1994. [ Links ]
23. Brown, R. F. C.; Donohue, A. C.; Jackson, W. R.; McCarthy, T. D. Tetrahedron 1994, 50, 13739-13752. [ Links ]
24. CCDC (Cambridge Crystallographic Data Centre) numbers, 158726, 158727 and 158868 for compounds 1, 2 and 5, respectively.

