




![Síntesis y caracterización de oligosilanos complejos en el sistema pentametilciclopentadienil dicarbonilo de fierro: [η5-C5(CH3)5]Fe(CO)2Si n(Si n = SiMe3; Si2Me5; Si3Me7; y 2-Si3Me7)](/img/es/next.gif)





Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.44 no.2 Ciudad de México abr./jun. 2000
Investigación
New Palladium(II) Tetraorganodichalcogenoimido Diphosphinates. Crystal and Molecular Structure of Pd[(SPMe2)2N]2 and cis-Pd[(OPPh2)(SPMe2)N]2
Danut Bilc,1 Anca Silvestru,1 Cristian Silvestru,1 Ionel Haiduc,1* and John E. Drake2*
1 Facultatea de Chimie, Universitatea Babes-Bolyai, RO-3400 Cluj-Napoca, Romania.
2 Department of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario, Canada, N9B 3P4.
Recibido el 18 de enero del 2000.
Aceptado el 23 de marzo del 2000.
Abstract
Bis(tetraorganodichalcogenoimidodiphosphinato)palladium(II), Pd[(XPR2)(YPR'2)N]2, were prepared and investigated by means of IR and multinuclear (1H, 13C, 31P) NMR spectroscopy. The crystal and molecular structures of Pd[(SPMe2)2N]2 (1) and cis-Pd [(OPPh2)(SPMe2)N]2 (2) were determined by X-ray diffractometry. The crystals of both compounds contain discrete mononuclear molecules. The ligands act as monometallic biconnective units through both chalcogen atoms which results in a spiro-bicyclic system with distorted square planar PdS4 and PdS2O2 core, respectively.
Key Words. Bis(tetraorganodichalcogenoimidodiphosphinato)palladium(II), X-ray diffractometry, monometallic biconnective units, spiro-bicyclic system.
Resumen
Bis(tetraorganodichalcogenoimidodifosfinato) de paladio (II), Pd[(XPR2)(YPR'2)N]2 fueron preparados e investigados por espectroscopía de infrarrojo y resonancia magnética multinuclear (1H, 13C, 31P). Las estructuras moleculares y cristalinas de Pd[(SPMe2)2N]2 (1) y cis-Pd[(OPPh2)(SPMe2)N]2 (2) fueron determinadas por difracción de rayos X. Los cristales de ambos compuestos contienen moléculas mononucleares discretas. Los ligantes actúan como unidades biconectivas monometálicas a traves de ambos átomos chalcogénicos resultando un sistema espiro bicíclico con un núcleo planar cuadrado distorsionado, PdS4 y PdS2O2, respectivamente.
Palabras clave: Bis(tetraorganodichalcogenoimidodifosfinato) de paladio (II), difracción de rayos X, unidades biconectivas monometálicas, sistema espiro bicíclico.
Dedicated to the memory of Dr. Jacobo Gómez-Lara, a remarkable man, scientist and friend
Introduction
The synthesis of several Pd(II) complexes of tetraorganodichalcogenoimidodiphosphinato ligands has been described in the literature [1-14] and the molecular structure of either neutral or anionic compounds has been established by X-ray diffractometry. Most of the known bis(tetraorganodichalcogenoimidodiphosphinato)palladium(II) complexes contain symmetric organophosphorus moieties, Pd[(XPR2)2N]2 [1-5,13]. Few complexes of asymmetric ligands, i.e. different chalcogen atoms and/or organic groups attached to phosphorus atoms, Pd[(XPR2) (YPR'2)N]2, have been described so far: Pd[(SP Ph 2){SP (OEt)2}N]2 [6], Pd[(OPPh2)(YPPh2)N]2, Pd[(OPPh2)(YPPh2) N]2(en) (Y = S, Se) [9,10], Pd[(OPPh2){SP(OEt)2} N]2, Pd[(SPPh2){OP(OEt)2}N]2 [11], Pd[{OP (OPh)2}{SP (OPh)2} N]2 [14]. Regardless of the nature of the organophosphorus ligands, with one exception, both chalcogen atoms are involved in coordination to the metal center (a), and this results in a square planar coordination geometry for the bis (tetraorganodichalcogenoimido-diphosphinato)palladium(II) complexes.
When asymmetric ligands have been used, NMR spectroscopic evidence has suggested in some cases that both cis (b) and trans (c) isomers are present in solution [6,11]. For Pd[(SPPh2){SP(OEt)2}N]2, the trans-isomer (with respect to the organic groups attached to phosphorus atoms) was isolated in the solid state [6], while only the cis isomers have been isolated and characterized by single crystal X-ray diffractometry for Pd[(OPR2)(YPR'2)N]2 (Y = S, Se). Only for the Pd[{OP (OPh)2}{SP(OPh)2}N]2 complex was a different molecular structure found; in this case the organophosphorus ligands exhibit a S,N-monometallic biconnective coordination pattern, resulting in a trans-NiS2N2 square planar core (e) [14]. When ethylenediamine was used as a chelating agent the [(OPPh2) (YPPh2)N]- (Y = S, Se) ligands were found to exhibit a monometallic monoconnective coordination pattern only through the "soft" chalcogen atom (d) [9,10].
We report here on the synthesis and spectroscopic characterization of new Pd[(XPR2)(YPR'2)N]2 complexes, and the crystal and molecular structures of Pd[(SPMe2)2N]2 and cis-Pd[(OPPh2)(SPMe2)N]2.
Results and Discussion
The title compounds were prepared as red-orange to red-brown crystalline solids, by reacting palladium dichloride, PdCl2, with the appropriate sodium or potasium salts of the tetraorganodichalcogenoimidodiphosphinic acids in acetone, according to equation (1):
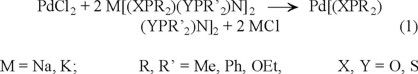

Details concerning the preparation, including yields and melting points, are given in the Experimental Section. All compounds were recrystallized by slow diffusion of n-hexane into an acetone solution of the Pd(II) complex.
The new Pd(II) complexes were investigated by IR, and multinuclear (1H, 13C, 31P) NMR spectroscopy, and the crystal and molecular structures of Pd[(SPMe2)2N]2 and cis-Pd [(OPPh2)(SPMe2)N]2 were determined by X-ray diffractometry.
The strong IR absorptions observed for the Pd(II) complexes in the regions 1250-1150, 1070-1050 and 560-510 cm−1 were assigned to νas(P2N), ν(PO), and ν(PS) stretching vibrations, respectively, by comparison with the spectra of the free acids and their alkali metal salts. This behavior is consistent with the coordination of the ligands in the deprotonated form through both chalcogens to the metal atom.
1H and 13C NMR spectra show characteristic signals for the organic groups attached to phosphorus, with the expected pattern due to phosphorus-proton/carbon couplings. Thus, for example, the 1H NMR spectrum of Pd[(OPPh2){OP(OEt)2}N]2 exhibits two resonances with the expected splitting pattern for P-OCH2CH3 (triplet) and P-OCH2CH3 (doublet of quartets) in the aliphatic region, and two resonances (P-C6H5-ortho - doublet of doublets, and P-C6H5-meta+para - multiplet) in the aromatic region, respectively. This indicates the equivalence of the organic groups attached to the same phosphorus atom on the NMR time-scale.
The 31P NMR spectra of compounds 1 - 3 are of the typical AX type, thus indicating the presence of only one isomer in solution for complexes 2 and 3. By contrast, for Pd[(SP Me2)(SPPh2)N]2 (4) two AX spectra with very similar chemical shifts were observed. This behavior is most likely due to the presence of both cis and trans isomers in solution. All four resonances exhibit a doublet pattern due to the phosphorus-phosphorus coupling. Similar behavior was previously described for the Pd[(SPPh2){SP(OEt)2}N]2 complex [6]. The 31P NMR spectra are consistent with a coordination pattern of the tetraorganodichalcogenoimidodiphosphinato ligands through both chalcogens to the metal atom.
Crystal and molecular structure of Pd[(SPMe2)2N]2 (1) and cis-Pd[(OPPh2)(SPMe2)N]2 (2)
The solid state structures of Pd[(SPMe2)2N]2 (1) and cis-Pd[(OPPh2)(SPMe2)N]2 (2) were determined by single-crystal X-ray diffraction. For both compounds the crystal contains discrete monomeric molecules separated by normal van der Waals distances. The ORTEP plots of the molecular structures of 1 and 2 are displayed in Fig. 1 and 2. Important bond lengths and angles are listed in Table 1.
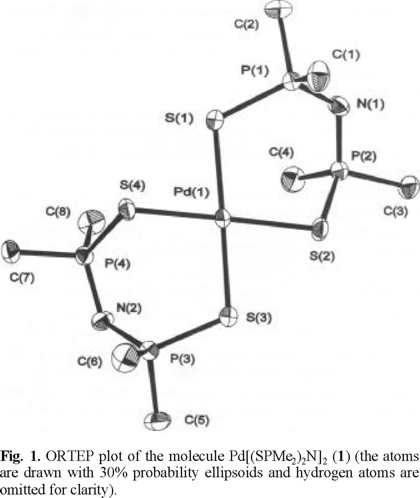
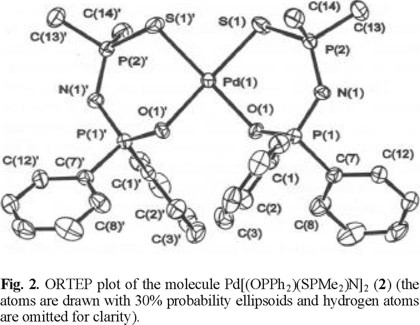
The molecular structure of 1 and 2 complexes contains monometallic biconnective tetraorganodichalcogenoimidodi-phosphinato ligands, coordinated to the metal center through both chalcogen atoms. The result is a spiro-bicyclic system with a square-planar PdS2X2 core slightly distorted to tetrahedral [dihedral angle Pd(1)S(1)S(2) / Pd(1)S(3)S(4) 8.4° in 1, and Pd(1)O(1)S(1) / Pd(1)O(1a)S(1a) 3.9° in 2, respectively], and a cis arrangement of the donor atoms around the palladium center in the monothioimidodiphosphinato complex 2, as observed for other Pd[(OPR2)(YPR'2)N]2 (Y = S, Se) derivatives [10,11].
The Pd-S [range 2.332(1)-2.353(1) Å in 1, and 2.292(1) Å in 2] bond distances are very close to those found in the related palladium(II) derivatives, e.g. Pd[(SPPh2){SP(OEt)2} N]2 (av. 2.335 Å) [6], Pd[{SP(OPh)2}2N]2 (av. 2.345 Å) [13], or Pd[{OP(OPh)2}{SP(OPh)2}N]2 (av. 2.342 Å) [14], while the Pd-O [2.071(3) Å] bond distances in 2 are similar to those found in Pd[(OPPh2)(SePPh2)N]2 (av. 2.080 Å) [10].
The P-S [range 2.028(1)-2.036(1) Å in 1, and 2.042(2) Å in 2] and the P-N bonds [range 1.591(3)-1.600(3) Å in 1, and 1.580(4)-1.593(4) Å in 2] are equal, within the experimental errors, and their lengths are intermediate between single and double phosphorus-sulphur and phosphorus-nitrogen bonds, cf. the free acids (S=PMe2)2NH [15]: P=S 1.939(2), 1.962(2), P-N 1.679(3), 1.675(3) Å; (O=PPh2)(S=PMe2)NH [16]: P=S 1.944(3), (S)P-N 1.681(7), (O)P-N 1.662(7), P=O 1.480(5) Å; Ph2P(S)SH [17]: P=S 1.954(1), P-S 2.077(1) Å; and [(Me3Si)2N-P(=NBut)S]2 [18]: P=N 1.529(2), P-N 1.662(2) Å. The P-O [1.512(3) Å in 2] bond distances have also intermediate values between single and double phosphorus-oxygen bonds (cf. Ph2P(=O)OH [19]: P-O 1.526(6), P=O 1.486(6) Å), and are comparable with those observed in cis-Pd[(OPPh2)(SePPh2)N-O,Se]2 (av. P-O 1.521 Å) [10]. In the trans-Pd[{OP(OPh)2}{SP(OPh)2}N]2 complex, containing S,N-coordinated ligands, the corresponding P=O bond distance is significantly shorter [1.450(3) Å] [14].
The six-membered PdSXP2N rings in both Pd(II) complexes are not planar and their conformation, as reflected by the torsion angles (Table 2), is consistent with a high flexibility of the SPNPX systems. Thus, for 1 the two chelate rings display twisted boat conformations, with P(1)/S(2) and P(3)/ S(4) atoms, respectively, in the apices, and are bent on the same side of the PdS4 core of the spiro-bicyclic NP2S2PdS2 P2N system [deviations from the best PdS4 plane: N(1) 0.414, N(2) 1.679 Å]. By contrast, in 2 the chelate rings display twisted chair conformations, with Pd(1)/N atoms in the apices, and they are oriented on opposite sides of the PdS2O2 core of the cis spiro-bicyclic NP2SOPdOSP2N system [deviations from the best PdS4 plane: N(1) -1.779, N(1a) 1.779 Å].
Experimental
Sodium or potassium tetraorganodichalcogenoimidodiphosphinates were prepared according to published methods: K[(SPMe2)2N] [15], K[(OPPh2)(SPMe2)N] [16], K[(OPPh2) {OP(OEt)2}N] [20], K[(SPMe2)(SPPh2)N] [21]. Palladium dichloride, PdCl2, was commercially available. The infrared spectra were recorded using KBr pellets on a Specord 75 IR C. Zeiss-Jena (DDR) instrument, in the range 4000-400 cm−1. 1H, 13C and 31P NMR spectra were recorded on a Varian Gemini 300S instrument operating at 299.5, 75.4 and 121.4 MHz, respectively. The chemical shifts are reported in ppm relative to TMS and H3PO4 85%, respectively.
Preparation of the title compounds, Pd[(XPR2)(YPR'2)N]2, (Table 3)
A reaction mixture containing stoichiometric amounts of PdCl2 and the corresponding sodium or potasium tetraorganodichalcogenoimidodiphosphinate, M[(XPR2)(YPR'2)N], in 40 mL acetone was stirred at room temperature for 24 h. The insoluble solid was filtered off and from the clear dark-red solution the solvent was removed to dryness in a rotary evaporator. The remaining solid product was recrystallized from organic solvents.
Pd[(SPMe2)2N]2 (1): 1H NMR (CDCl3, 300 MHz) 1.92 (m, 2JPH + 4JPH 12.2 Hz); 13C NMR (CDCl3, 75 MHz) 25.8 (m, 1JPC + 3JPC 77.3 Hz); 31P NMR (CDCl3, 121,4 MHz) 41.1 (s). Pd[(OPPh2)(SPMe2)N]2 (2): 1H NMR (CDCl3, 300 MHz) 1.55 (d, 3H, 2JPH 14.1 Hz, CH3), 7.50 (m, 3H, C6H5 - meta + para), 7.83 (dd, 2H, 3JPH 12.9, 3JHH 7.8 Hz, C6H5 - orto); 13C NMR (CDCl3, 75.4 MHz) 25.38dd (dd, 1JPC 67.8, 3JPC 6.0 Hz, CH3), 128.76 (d, 3JPC 12.2 Hz, Cm), 131.46 (s, Cp), 132.04 (d, 2JPC 9.5 Hz, Co) (ipso-carbons not observed); 31P NMR (CDCl3, 121.4 MHz) 22.6 (d, 2JPP 8.5 Hz, Ph2PO), 38.1 (d, 2JPP 8.5 Hz, Me2PS).
Pd[(OPPh2){OP(OEt)2}N]2 (3): 1H NMR (Me2CO-d6, 300 MHz) 1.06 (t, 3H, 3JHH 7.1 Hz, P-OCH2CH3), 3.88 (dq, 2H, 3JPH 6.9, 3JHH 6.9 Hz, P-OC H2CH3), 7.29 (m, 3H, P-C6H5-meta+para), 7.96 (dd, 2H, 3JHH 7.9, 3JPH 11.8 Hz, P-C6H5-orto); 13C NMR (Me2CO-d6, 75.4 MHz) 16.54 (d, 3JPC 8.0 Hz, P-OCH2CH3), 61.61 (d, 2JPC 4.8 Hz, P-OCH2CH3), 128.31 (d, 3JPC 12.7 Hz, Cmeta), 130.26 (s, Cpara), 132.15 (d, 2JPC 9.8 Hz, Corto), 141.19 (dd, 1JPC 131.7, 3JPC 4.3 Hz, Cipso); 31P NMR (Me2CO-d6, 121.4 MHz) 3.0 (d, 2JPP 20.9 Hz, EtOPO), 13.3 (s, Ph2PO).
Pd[(SPMe2)(SPPh2)N]2 (4): 31P NMR (CDCl3, 121.4 MHz) 37.15 (d, 2JPP 4.5 Hz, Ph2PS), 37.76 (d, 2JPP 4.5 Hz, Ph2PS), 43.64 (d, 2JPP 4.5 Hz, Me2PS), 43.70 (d, 2JPP 4.5 Hz, Me2PS) (see text).
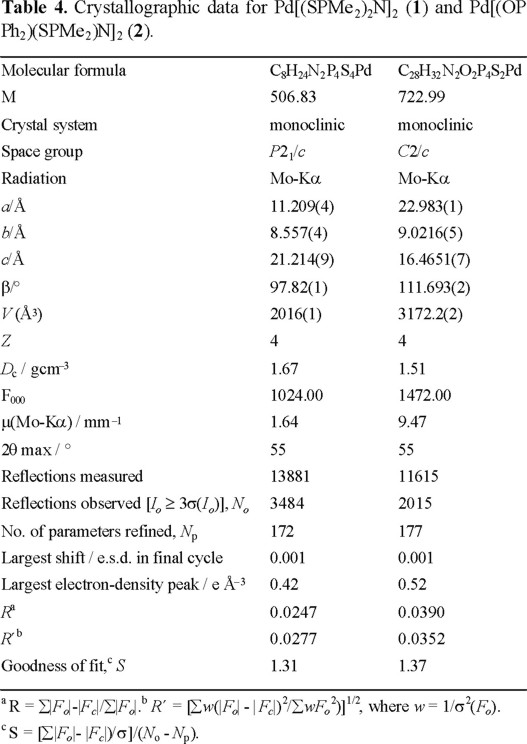
Crystal structure determinations. A red block crystal of Pd[(SPMe2)2N]2, 1, and an orange block crystal of Pd[(OP Ph2)(SPMe2)N], 2 were mounted on glass fibres and sealed with epoxy glue. Data were collected on a Siemens SMART/CCD system at McMaster University with graphite-monochromated Mo-Ka radiation, operating at 50 kV and 35 mA. Cell constants corresponded to monoclinic cells whose dimensions are given in Table 4 along with other experimental parameters. Semiempirical absorption corrections were applied which resulted in transmission factors ranging from 0.831 to 0.696 for 1, and 0.883 to 0.713 for 2. The structures were solved by direct methods [22]. All of the non-hydrogen atoms were treated anisotropically. Hydrogen atoms were included in idealized positions with C-H set at 0.95 Å and with isotropic thermal parameters set at 1.2 times that of the carbon atom to which they were attached. The final cycle of full-matrix least-squares refinements [23] were based on 3484 (for 1) and 2015 (for 2) observed reflections [I > 3.00σ(I)] and 172 (for 1) and 177 (for 2) variable parameters and converged (largest parameter shift was 0.001 times its esd) with unweighted and weighted agreement factors of R = ∑|Fo|-|Fc|∑|Fo| = 0.0247 (for 1) and 0.0390 (for 2) and R´ = |∑w(|Fo|-|Fc|)2/∑wFo2)]1/2 = 0.0277 (for 1) and 0.0352 (for 2). The standard deviations of an observation of unit weight [24] was 1.31 (for 1) and 1.37 (for 2). Plots of [∑w(|Fo|-|Fc|)2 versus |Fo|, reflection order in data collection, sin ϑ/λ, and various classes of indices showed no unusual trends. The maximum and minimum peaks on the final difference Fourier map corresponded to 0.42 and −0.68 e/Å3, respectively (for 1) and 0.52 and −0.84 (for 2). Neutral-atom scattering factors were taken from Cromer and Waber [25]. Anomalous dispersion effects were included in Fc [26]; the values for Δf ' and Δf" were those of Creagh and McAuley [27]. All calculations were performed using the TEXAN TEXSAN [28] crystallographic package of Molecular Structure Corp. Selected distances and bond angles are given in Table 1 and the molecule is displayed in the ORTEP diagram in Figures 1 and 2. Additional material available from the Cambridge Crystallographic Data Centre comprises the final atomic coordinates for all atoms, thermal parameters, and a complete listing of bond distances and angles.
Supplementary material. The complete listing of the crystal data is provided in the Supporting Information which contains tables giving final fractional coordinates and B(eq) for non-hydrogen atoms, anisotropic thermal parameters for non-hydrogen atoms, atomic coordinates and B(eq) for hydrogen atoms, all bond lengths and angles and ORTEP drawings for Pd[(SPMe2)2N]2 and Pd[(OPPh2)(SPMe2)N]2 (12 pages). Structure-factor tables are available from the authors (J.E.D.).
Acknowledgements
This work was supported by the Romanian Academy and the Romanian National Council for High Education Scientific Research (CNCSIS) and by the Natural Sciences and Engineering Research Council of Canada. We also thank Jim Britten of the McMaster Chemistry X-ray Facility for providing help and facilities when they became unavailable to one of us (J.E.D.) at the University of Windsor.
References
1. Bhattacharyya, P.; Novosad, J.; Phillips, J.; Slawin, A. M. Z.; Williams, D. J.; Woollins, J. D. J. Chem. Soc., Dalton Trans. 1995, 1607-1613. [ Links ]
2. Cupertino, D.; Keyte, R. W.; Slawin, A. M. Z.; Williams, D. J.; Woollins, J. D. Phosphorus, Sulfur & Silicon 1996, 109-110, 193-196. [ Links ]
3. Cupertino, D.; Keyte, R. W.; Slawin, A. M. Z.; Woollins, J. D.; Williams, D. J. Polyhedron 1996, 15, 4441-4445. [ Links ]
4. Papadimitriou, C.; Veltsistas, P.; Novosad, J.; Cea-Olivares, R.; Toscano, A.; Garcia y Garcia, P.; López-Cardoso, M.; Slawin, A. M. Z.; Woollins, J. D. Polyhedron 1996, 16, 2727-2729. [ Links ]
5. Cupertino, D.; Keyte, R. W.; Slawin, A. M. Z.; Woollins, J. D. Polyhedron 1998, 18, 707-716. [ Links ]
6. Cupertino, D.; Keyte, R. W.; Slawin, A. M. Z.; Woollins, J. D. Polyhedron 1998, 18, 311-319. [ Links ]
7. Bhattacharyya, P.; Slawin, A. M. Z.; Martin, M. B. J. Chem. Soc., Dalton Trans. 1998, 2467-2475. [ Links ]
8. Bhattacharyya, P.; Slawin, A. M. Z.; Williams, D. J.; Woollins, J. D. J. Chem. Soc., Dalton Trans. 1995, 2489-2495. [ Links ]
9. Slawin, A. M. Z.; Martin, M. B.; Woollins, J. D. J. Chem. Soc., Dalton Trans. 1998, 1537-1539. [ Links ]
10. Slawin, A. M. Z.; Martin, M. B.; Woollins, J. D. J. Chem. Soc., Dalton Trans. 1996, 3659-3665. [ Links ]
11. Cupertino, D.; Keyte, R. W.; Slawin, A. M. Z.; Woollins, J. D. Polyhedron 1998, 17, 4219-4226. [ Links ]
12. Slawin, A. M. Z.; Martin, M. B.; Woollins, J. D. Polyhedron 1998, 17, 4465-4473. [ Links ]
13. Nouaman, M.; Zak, Z.; Herrmann, E.; Navratil, O. Z. Anorg. Allg. Chem. 1993, 619, 1147-1153. [ Links ]
14. Zak, Z.; Fofana, M.; Kamenicek, J.; Glowiak, T. Acta Crystallogr. 1989, C45, 1686-1689. [ Links ]
15. Silvestru, C.; Rösler, R.; Haiduc, I.; Cea-Olivares, R.; Espinosa-Perez, G. Inorg. Chem. 1995, 34, 3352-3354. [ Links ]
16. Rösler, R.; Stanciu, M.; Yang, J.; Drake, J. E.; Silvestru, C.; Haiduc, I. Phosphorus, Sulfur & Silicon 1998, 132, 231-250. [ Links ]
17. Krebs, B.; Henkel, Z. Z. Anorg. Allg. Chem. 1981, 475, 143-155. [ Links ]
18. Pohl, S. Chem. Ber. 1976, 109, 3122- [ Links ].
19. Fenske, D.; Mattes, R.; Lons, J.; Tebbe, K.-F. Chem. Ber. 1973, 106, 1139-1144. [ Links ]
20. Balazs, G.; Silvestru, C.; Rösler, R.; Haiduc, I.; Cea-Olivares, R.; Espinosa-Perez, G. Inorg. Chim. Acta, 1999, 287, 61-71. [ Links ]
21. Silvestru, C.; Rösler, R.; Drake, J. E.; Yang, J.; Espinosa-Perez, G.; Haiduc, I. J. Chem. Soc., Dalton Trans. 1998, 73-78. [ Links ]
22. Sheldrick, G. M. Acta Crystallogr. 1990, A46, 467. [ Links ]
23. Least-squares: Function minimized: ∑w(|Fo| - |Fc|)2, where w = 4Fo2(Fo2), σ2(Fo2) = [S2(C + R2B) + (pFo2)2]/(Lp)2, S = scan rate, C = total integrated peak count, R = ratio of scan time to background counting time, Lp = Lorentz-polarization factor, and p = p factor.
24. Standard deviation of an observation of unit weight: [∑w(|Fo| -|Fc|)2/No - Nv)1/2, where No = number of observations and Nv = number of variables.
25. Cromer, D. T.; Waber, J. T. International Tables for X-ray Crystallography, Kynoch Press, Birmingham, 1974, Vol. 4, Table 2.2A. [ Links ]
26. Ibers, J. A.; Hamilton, W. C. Acta Crystallogr. 1964, 17, 781-784. [ Links ]
27.Creagh, D. C.; McAuley, W. J. International Tables for Crystallography, Kluwer Academic Publishers, Boston, U.S.A., 1992, Vol. C, Table 4.2.6.8. [ Links ]
28. TEXSAN-TEXRAY Structure Analysis Package, Molecular Structure Corporation, Woodlands, TX, 1985 and 1992. [ Links ]

