










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.44 n.2 Ciudad de México Apr./Jun. 2000
Investigación
A New Approach for the Synthesis of the Cyclohexene Core of Anthracyclines and Milbemycins
Raúl Aguilar, Alicia Reyes, Arturo Orduña, Gerardo Zepeda, Roderick W. Bates,‡ and Joaquín Tamariz*
Departamento de Química Orgánica, Escuela Nacional de Ciencias Biológicas, I.P.N. Prol. Carpio y Plan de Ayala, 11340 México, D.F. Phone: (52)-5729-6300/62411; Fax: (52)-5396-3503. E-mail: jtamariz@woodward.encb.ipn.mx
‡ Department of Chemistry, Chulalongkorn University, Phaya Thai Road, Bangkok 10330, Thailand.
Recibido el 6 de diciembre de 1999.
Aceptado el 28 de enero del 2000.
Abstract
A new synthesis of the cyclohexene core of the anthracyclines and milbemycins antibiotics is described, using 3-p-nitroben-zoyloxy-3-buten-2-one (9a) as an efficient dienophile in Diels-Alder reactions with substituted dienes. Allylic functionalization and epimerization of the cycloadducts led, in moderate overall yields, to the corresponding related cyclohexene A-rings of aclacinomycin, α- and β-rhodomycins, and the cyclohexene moiety of milbemycins β1 and E.
Key Words: Captodative olefin, Diels-Alder, cyclohexene core, anthracyclines, milbemycins.
Resumen
Se describe una nueva ruta sintética para la preparación de la unidad ciclohexénica de algunos antibióticos de la familia de las antraciclinas y milbemicinas, empleando la 3-p-nitrobenzoiloxi-3-buten-2-ona (9a) como dienófilo en reacciones de Diels-Alder con dienos sustituidos. La funcionalización alílica y epimerización de los aductos condujo en rendimientos moderados a anillos ciclohexénicos análogos a los correspondientes de la aclacinomicina, de las rodomicinas alfa y beta, y también del fragmento ciclohexénico de las milbemicinas β1 y E.
Palabras clave: Olefina captodativa, Diels-Alder, núcleo ciclohexénico, antraciclinas, milbemicinas.
Dedicated to the memory of Dr. Lydia Rodriguez-Hahn and Dr. Jacobo Gómez-Lara
Introduction
Anthracyclines are tetrahydronaphthacenequinone antibiotics isolated from numerous actinomycete species, and they are considered among the most active antineoplastic drugs [1]. Daunorubicin (1) and adriamycin (2) are clinically broad chemotherapeutic agents used for leukemia and cancer treatment [2]. Their structures are characterized by a tetracyclic aglycone to which an aminoglycoside is linked to the C-7 hydroxyl group. Aclacinomycin (3) is one of the most important member of the aklavinone antibiotics group [3]. Its major structural difference with respect to 1 and 2 is the methoxycarbonyl group at the C-10 position, and the absence of the carbonyl group at the C-9 side chain. Instead of this group on C-10, β-rhodomycin (4), a member of a large series of analogs, has a hydroxy group, and the ethyl substituent on C-9 is preserved [4]. Interestingly, a common structural feature in all these compounds is the quaternary carbon C-9 bearing an alpha hydroxy group.

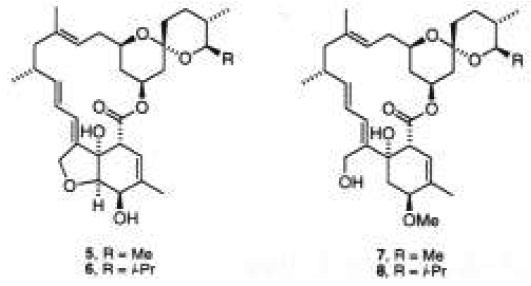
A further relevant class of antibiotics are avermectins and milbemycins, which are sixteen-membered ring macrolides isolated from Streptomyces hygroscopicus [5]. They have shown a potent and broad activity spectrum as anthelmintics, acaricides, and insecticides. Some of them have been commercialized for agricultural and veterinarian purposes, such as milbemycin α1 (5) and D (6) [6]. In contrast, milbemycins β1 (7) and E (8), among other natural analogs, do not have the tetrahydrofuran ring fused to the highly functionalized cyclohexene moiety [6b]. Consequently, an intense effort has been devoted to the synthesis of these important molecules [6, 7].
Previously, we have demonstrated that captodative olefins 3-aroyloxy-2-butenones 9 were highly regio- and stereo-selective in Diels-Alder cycloadditions towards unsymmetrical carbocyclic dienes [8], and they proved to be efficient synthons in the synthesis of natural terpenoids [9]. Considering that these olefins are able to introduce an alpha-ketol on a quaternary carbon, like the cyclohexene moiety of anthracyclinones 1-2 and potentially of 3-4, and milbemycins 7 and 8, we have undertaken a study for the preparation of the cyclohexenic core of these molecules via a Diels-Alder addition of captodative olefin 9a to the suitable dienes, followed by functionalization of the corresponding adducts (Fig. 1).
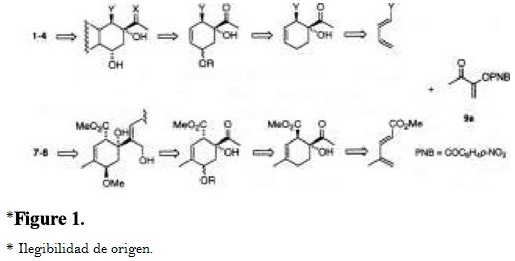
Results and Discussion
Thermal reaction (xylene, 130 °C, 11 h) of 1-acetoxy-1,3-butadiene (10a) with olefin 9a provided adduct 11a as a single isomer (Fig. 2) [8a]. In contrast, the cycloaddition of 9a with 1-(methoxycarbonyl)-1,3-butadiene (10b), under similar conditions, yielded a mixture of stereoisomers 11b and 12b (80:20) in good yield (81%). The major isomer was isolated from the reaction mixture as colorless needles by recrystallization (hexane/EtOAc, 8:2). No regioisomers 13 were detected by NMR in the crude mixtures [8a].

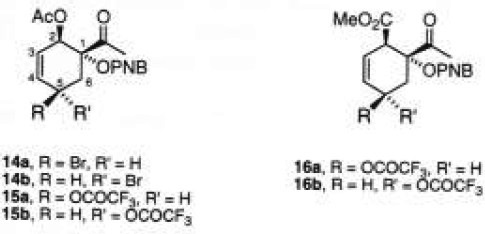
Previously, allylic hydroxylation of cyclohexenes has been successfully attained by treatment with NBS in two phases (CCl4/H2O) [10]. This method was applied on adduct 11a giving a complex mixture of products, including the aromatized acetophenone product. Other reagents for the allylic oxidation such as CrO3 and SeO2 were also used [11], yielding no conversion. The best results were found by non-aqueous bromination (NBS, CH2Cl2/CCl4, 8:2, 50 °C, 30 min), and succesive trifluoroacetoxylation by adding silver trifluoroacetate in ether, furnishing a mixture of epimers 15a/15b (60:40) in 29% yield [12]. It seems that the bromination step was more efficient, since we were able to isolate the bromocyclohexenes 14a/14b in 60% yield as an unstable mixture, which undergoes elimination and aromatization. Analogous results were obtained with adduct 11b. Thus, under the same conditions of allylic trifluoroacetoxylation, the expected products 16a/16b (60:40) were isolated with a yield (30%) comparable to that observed for derivatives 15. The isomeric ratios of 15 and 16 were calculated from the 1H NMR spectra, but the configuration attributed to carbon C-5 was not unambiguously established.
Therefore, this methodology gives rise to diasteroisomers 15a and 15b, which are equivalent synthons of the A-rings of α- and β-rhodomycins, 17 and 4, respectively (Fig. 3) [4]. Inasmuch as one wishes to separate isomers 15a and 15b, the major compound 15a would be, for the best, obtained in 60% yield. On the other hand, in spite of the unfavorable ratio of 15b for the synthesis of the A-ring of 4, the epimerization of carbon C-5 under acidic conditions may afford the most stable alpha epimer 4 [13].
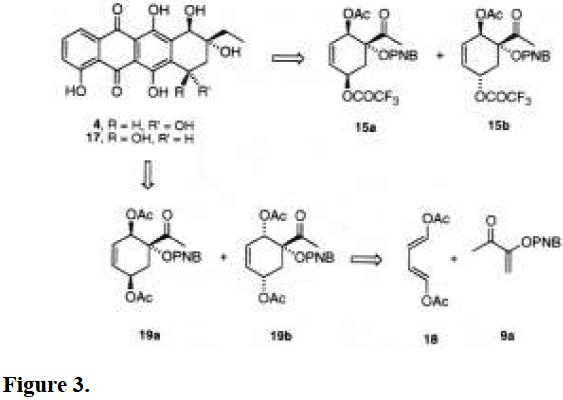
A more convergent synthesis of the A-ring of α-rhodomycin (17) may be designed by cycloaddition of olefin 9a with the diene (E,E )-1,4-diacetoxy-1,3-butadiene (18), which is already bearing the oxygenated functionalities (Fig. 3). The thermal (xylene, 130 °C, 16 h) addition of these starting materials yielded a mixture of adducts 19a/19b (9:1), as previously reported [8a].
We have explored the hydrolysis of 19a with the aim to prepare a derivative structurally closer to the A-ring of 17. When 19a was treated with potassium carbonate in dry methanol at 25 °C for 2 h, a single product was obtained in high yield (92%). Double irradiation experiments in 1H NMR and 13C NMR spectra showed that the structure would be attributed to diol 20 (Fig. 4). Longer reaction times led to epimerization of carbon C-2, before the hydrolysis of the third acetate takes place. Further efficient conditions to prepare compound 21 are currently being investigated.

In order to approach the synthesis of the cyclohexene moiety of milbemycins β1 (7) and E (8) (Fig. 1), diene 24 was prepared by the Wittig-type reaction of methacrolein (22) with methyl diethylphosphonoacetate (23) (Fig. 5) [14]. The thermal addition of diene 24 with olefin 9a was highly stereo- and regio-selective, giving almost exclusively the endo and ortho-adduct 25 in good yield (70%) (Table 1).

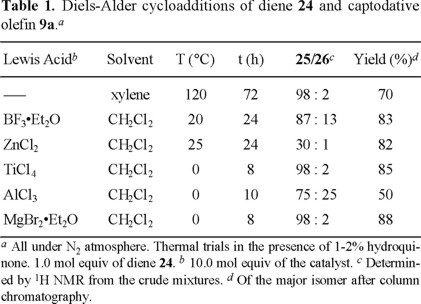
Several Lewis acids were tested in order to improve the reaction conditions and yields (Table 1). The most efficient catalysts were ZnCl2, TiCl4, and MgBr2•Et2O, since the stereoselectivity was as high as the thermal trial. Moreover, with the last two Lewis acids, the reaction temperature and time were the lowest.
The preference for the endo selectivity and regioselectivity agrees with that observed for diene 10b. The electronic factors at the transition state which control the selectivity also seem to be involved with diene 24 [8a]. In addition, the presence of the methyl group in 24 should enhance the regioselectivity.
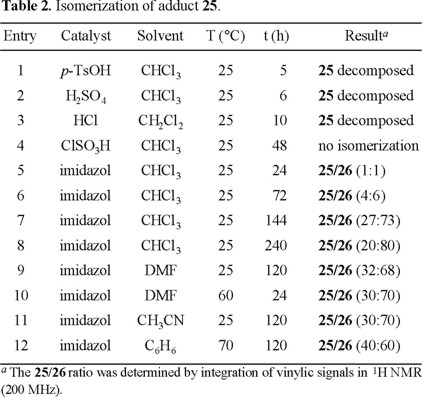
Nevertheless, the major isomer 25 does not have the required relative configuration on carbons C-1 and C-2, and it should be isomerized to epimer 26 under kinetically controlled acidic or basic conditions. Indeed, when 25 was treated with imidazole in chloroform at room temperature for 24 h, a mixture of 25/26 (1:1) was obtained, and after stirring for 10 days, this ratio reached up to 25/26 (2:8) (Table 2, entries 5-12). A favorable ratio was reached faster by using more polar solvents such as DMF and acetonitrile. This isomerization was unsuccessful under acidic conditions with diverse catalysts (p-TsOH, H2SO4, HCl, and ClSO3H).
The methanolysis of adduct 25 with potassium carbonate in dry MeOH was carefully studied, since an alternative product was generated when the reaction conditions were not properly maintained. Ketol 27 was isolated in 55% yield after stirring the mixture in THF with MeOH (25 mol eq) at 0 °C for 72 h, and extracting the crude with EtOAc followed by cold washings with aqueous saturated solution of NH4Cl until neutral, and drying with anhydrous Na2SO4. If this procedure is not carefully followed, ketol 27 is obtained in low yield, and a new compound is produced as the main component of the reaction mixture. The usual spectroscopic analyses (IR, 1H and 13C NMR) and elemental analysis provided enough information for assigning the structure 28 to this compound. This new bicyclic lactone was very stable, in spite of the usually low stability of a dienic moiety. An optimized procedure for the preparation of 28 consisted in the treatment of 25 with a high excess of MeOH (250 mol eq) at room temperature for 48 h, which provided 65% of the desired product.

The formation of the bicyclic lactone 28 may be explained by the mechanism depicted in Fig. 6. The syn relative configuration of the acetyl group on carbon C-1, and the methoxycarbonyl group on carbon C-2 of adduct 25, allows an easy intramolecular transesterification and lactonization via intermediates 29 and 30, to give precursor 31. The cyclization process would be favored by the stability of the five-membered ring. It is likely that the dehydration step may be the last process due to the fact that α-hydroxy ketones are relatively stable to basic and acidic conditions. Therefore, after lactonization, the elimination of the hydroxy group in the beta position to the γ-lactone carbonyl group in the intermediate 32 will be favored by the formation of the very stable γ-butenolide system of 28. This would be also stabilized by the conjugated cyclohexenyl dienic skeleton.

In summary, we have demonstrated the synthetic potential of captodative olefin 9a as a valuable synthon for the preparation of cyclohexene rings properly functionalized. This methodology may also be considered as a selective approach toward the preparation of the A rings of the aglycones of aclacynomicin (3), α-rhodomycin (17), and β-rhodomycin (4), and the cyclohexene moiety of milbemycins β1 (7) and E (8).
Experimental
General. Melting points (uncorrected) were determined with an Electrothermal capillary melting point apparatus. IR spectra were recorded on a Perkin-Elmer 1600 spectrophotometer. 1H and 13C NMR spectra were obtained on a Varian EM-390 (90 MHz), Varian Gemini-300 (300 MHz), and Brucker DMX-200 (200 MHz) instruments, with CDCl3 as solvent and TMS as internal standard. The mass spectra (MS) were taken on a Hewlett-Packard 5971A spectrometer. Microanalyses were performed by M-H-W Laboratories (Phoenix, AZ). All air moisture sensitive reactions were carried out under nitrogen using oven-dried glassware. Benzene, toluene, and xylene were freshly distilled from sodium, and methylene chloride from calcium hydride, prior to use. K2CO3 was dried over-night at 120 °C before use. All other reagents were used without further purification. Compounds 9a, 11a, 11b, and 19a were prepared as described [8a, 9b].
[1R*,2R*,5R*]-1-Acetyl-2-acetoxy-5-trifluoroacetoxy-3-cyclohexen-1-yl p-Nitrobenzoate (15a). [1R*,2R*,5S*]-1-Acetyl-2-acetoxy-5-trifluoroacetoxy-3-cyclohexen-1-yl p-Nitro-benzoate (15b). To a solution of 0.10 g (0.29 mmol) of 11a in dry CH2Cl2 (1 mL) under an N2 atmosphere, and at 20 °C, 0.051 g (0.29 mmol) of NBS (recrystallized from benzene) in CCl4 (1 mL) were added. After being stirred at room temperature for 35 min under light irradiation (60 watts), CF3CO2Ag (0.06 g, 0.27 mmol) in dry ethyl ether (4 mL) was added, and the mixture was stirred and heated to 40 °C for 12.5 h. The mixture was diluted with CH2Cl2 (20 mL), filtered, and washed with cold water (3 × 5 mL). The organic layer was dried (Na2SO4) and the solvent was removed under vacuum. The residue was purified by column chromatography on Florisil (8 g, hexane/EtOAc, 8:2) to give 0.038 g (29%) of 15a/15b (60:40) as a colorless oil. 15a Rf 0.33 (hexane/EtOAc, 6:4); IR (film) 1740, 1720, 1710, 1530, 1365, 1300, 1240-1130, 800 cm−1; 1H NMR (200 MHz, CDCl3) δ 2.09 (s, 3H, CH3CO2), 2.24 (s, 3H, CH3CO), 2.50 (dd, J = 14.2, 7.6 Hz, 1H, H-6), 2.80 (dd, J = 14.2, 6.2 Hz, 1H, H-6), 4.18-4.24 (m, 1H, H-5), 5.62-5.65 (m, 1H, H-2), 5.82 (ddd, J = 10.1, 3.8, 1.7 Hz, 1H, H-3), 6.12 (ddd, J = 10.1, 2.2, 1.2 Hz, 1H, H-4), 8.08-8.17 (m, 2H, ArH), 8.28-8.33 (m, 2H, ArH). Signals attributed to the minor isomer 15b Rf 0.39 (hexane/EtOAc, 6:4): 1H NMR (90 MHz, CDCl3) δ 2.30 (s, CH3CO). Anal. Calcd for C19H16F3NO9: C, 49.68; H, 3.51. Found: C, 49.87; H, 3.28.
[1R*,2R*,5R*]-1-Acetyl-2-methoxycarbonyl-5-trifluoroacetoxy-3-cyclohexen-1-yl p-Nitrobenzoate (16a). [1R*,2R*, 5S*]-1-Acetyl-2-methoxycarbonyl-5-trifluoroacetoxy-3-cyclohexen-1-yl p-Nitrobenzoate (16b). The same procedure as for 15a/15b was used, with 0.15 g (0.43 mmol) of 11b, and 0.085 g (0.48 mmol) of NBS. The reaction was irradiated at 20 °C for 23 h, and 0.95 g (0.43 mmol) of CF3CO2Ag were added. The mixture was heated to 40 °C for 13 h. Column chromatography on Florisil (12 g, hexane/EtOAc, 6:4) yielded 0.04 g (30%) of 16a/16b (60:40) as a colorless oil: 16a Rf 0.21 (hexane/EtOAc, 6:4); IR (film) 1710, 1705, 1530, 1365, 1300, 1295, 1130, 760 cm−1; 1H NMR (90 MHz, CDCl3) δ 2.40 (s, 3H, CH3CO), 2.73-3.23 (m, 2H, H-6), 3.66 (s, 3H, CO2CH3), 3.68-5.77 (m, 1H, H-2), 4.19-4.28 (m, 1H, H-5), 5.79-5.88 (m, 1H, H-3), 6.02-6.12 (m, 1H, H-4), 8.08-8.17 (m, 2H, ArH), 8.26-8.35 (m, 2H, ArH). Signals attributed to the minor isomer 16b Rf 0.36 (hexane/EtOAc, 6:4): 1H NMR (90 MHz, CDCl3) δ 2.34 (s, CH3CO), 3.70 (s, 3H, CO2CH3), 4.65-4.73 (m, H-5); MS (70 eV) 163 (M+-296, 100), 147 (20), 120 (2), 105 (8), 77 (27), 75 (17).
[1R*,2R*,5R*]-1-Acetyl-5-acetoxy-1,2-dihydroxy-3-cyclohexene (20). To a solution of 0.5 g (1.23 mmol) of 19a in dry CH2Cl2 (6 mL) under an N2 atmosphere, and at 20 °C, 0.1 g (0.73 mmol) of K2CO3 in MeOH (2.5 mL) was added. After being stirred at room temperature for 2 h, the mixture was diluted with CH2Cl2 (20 mL), and washed with cold saturated solution of NH4Cl (3 × 5 mL) and cold brine until neutral. The organic layer was dried (Na2SO4) and the solvent was removed under vacuum. The residue was purified by column chromatography on silica gel (10 g, hexane/EtOAc, 9:1) to give 0.18 g (92%) of 20 as a dark yellow oil: Rf 0.24 (hexane/EtO Ac, 6:4); IR (film) 3450, 1715, 1705, 1540, 1345, 1310, 1100 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.05 (s, 3H, CH3CO2), 2.35 (s, 3H, CH3CO), 2.15-2.24 (m, 1H H-6β), 2.50-2.58 (m, 1H, H-6α), 4.07-4.13 (m, 1H, H-2), 4.50 (br, 2H, OH), 5.41-5.48 (m, 1H, H-5), 5.80-6.05 (m, 2H, H-3, H-4); 13C NMR (125 MHz, CDCl3) δ 21.2 (CH3CO2), 26.4 (CH3CO), 33.1 (C-6), 67.5 (C-2), 70.8 (C-5), 96.2 (C-1), 128.6 (C-3 or C-4), 130.7 (C-4 or C-3), 170.2 (CH3CO2), 211.0 (COMe).
Methyl (E)-4-methyl-2,4-pentadienoate (24) [15]. To a suspension of 0.510 g (0.012 mol) of LiCl in 60 mL of dry acetonitrile, under an N2 atmosphere and at room temperature, 2.18 g (0.012 mol) of trimethyl phosphonoacetate, 1.52 g (0.010 mol) of DBU, and 0.70 g (0.01 mol) of methacrolein were added dropwise. The mixture was stirred at room temperature for 24 h. The solvent was evaporated under vacuum, the residue was extracted with EtOAc (120 mL), and washed successively with 5% aqueous solution of HCl (2 × 40 mL), saturated solution of NaHCO3 (3 × 30 mL), and water until neutral. The organic layer was dried (Na2SO4) and the solvent was removed under vacuum. The residue was purified by distillation, using a Kugelrohr apparatus, to give 0.86 g (68%) of 24 as a pale yellow oil: Rf 0.92 (hexane/EtOAc, 7:3).
[1R*,2R*]-1-Acetyl-2-methoxycarbonyl-4-methyl-3-cyclohexen-1-yl p-Nitrobenzoate (25). Method A. A mixture of 0.10 g (0.42 mmol) of 9a, 0.159 g (1.26 mmol) of 24, and hydroquinone (3 mg) in dry xylene (2 mL), was placed in a threaded ACE glass pressure tube with a Teflon screw cap, under an N2 atmosphere, in the dark. The mixture was stirred and heated to 130 °C for 4 days. After cooling, it was diluted with EtOAc (60 mL) and washed with water (2 × 15 mL). The organic layer was dried (Na 2SO4), and the solvent was removed under vacuum. The residue was purified by column chromatography on silica gel (3 g, hexane/EtOAc, 8:2) to give 0.107 g (70%) of 25 as a pale yellow powder: Rf 0.89 (hexane/EtOAc, 7:3); mp 111-113 °C.
Method B. To a solution of 0.10 g (0.42 mmol) of 9a in dry CH2Cl2 (5 mL), under an N2 atmosphere at 0 °C, 0.779 g (4.10 mmol) of TiCl4, and 0.053 g (0.42 mmol) of 24 were added dropwise. The mixture was stirred for 8 h at 0 °C, diluted with EtOAc (60 mL), and washed successively with water (2 x 10 mL), saturated solution of NaHCO3 (3 × 15 mL), and water until neutral. The organic layer was dried (Na2SO4), and the solvent was removed under vacuum. The residue was purified by column chromatography on silica gel (3 g, hexane/EtO Ac, 8:2) to give 0.13 g (85%) of 25 as a pale yellow powder: IR (KBr) 2920, 1700, 1510, 1290 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.77 (s, 3H, Me-4), 1.90-2.08 (m, 1H, H-5α), 2.10-2.16 (dd, J = 18.0, 6.0 Hz, 1H, H-5β), 2.43 (s, 3H, CH3CO), 2.56 (ddd, J = 14.1, 11.4, 6.0 Hz, 1H, H-6β), 2.66 (dddd, J = 14.1, 6.3, 1.8, 1.5 Hz, 1H, H-6α), 3.69 (s, 3H, CO2CH3), 3.71-3.78 (m, 1H, H-2), 5.48-5.50 (m, 1H, H-3), 8.09-8.13 (m, 2H, ArH), 8.28-8.31 (m, 2H, ArH); MS (CI-NH3) 379 (M++18, 4), 362 (M++1, 6), 332 (20), 195 (100), 163 (13), 155 (18), 138 (18), 120 (21). Anal. Calcd for C18H19NO7: C, 59.83; H, 5.30. Found: C, 59.88; H, 5.40.
[1R*,2R*]-1-Acetyl-2-methoxycarbonyl-4-methyl-3-cyclohexen-1-ol (27). A solution of 0.20 g (0.55 mmol) of 25 in dry THF (5 mL), under an N2 atmosphere and at 0 °C, was treated with 0.911 g (6.60 mmol) of anhydrous K2CO3 in dry MeOH (0.306 mL, 13.7 mmol). After being stirred for 72 h at 0 °C, EtOAc (60 mL) was added, and the mixture was washed with aqueous 5% HCl (3 × 20 mL) and with cold water until neutral. The organic layer was dried (Na2SO4), and the solvent was removed under vacuum. The residue was purified by column chromatography on silica gel (6 g, hexane/EtOAc, 9:1) to give 0.064 g (55%) of 27 as a pale yellow oil: Rf 0.80 (hexane/EtOAc, 7:3); IR (CH2Cl2) 3455, 1709, 1439, 1355, 1271, 1197 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.74 (dd, J = 8.7, 3.7 Hz, 1H, H-6), 1.77 (br s, 3H, CH3C-4), 1.90-2.00 (m, 1H, H-5), 2.28-2.53 (m, 2H, H-5, H-6), 2.36 (s, 3H, CH3CO), 3.69 (s, 3H, CO2CH3), 3.79-3.85 (m, 1H, H-2), 4.18 (s, 1H, OH), 5.48-5.52 (m, 1H, H-3); 13C NMR (75 MHz, CDCl3) δ 23.3 (CH3), 24.0 (CH3CO), 25.7 (CH2), 30.9 (CH2), 47.5 (C-2), 52.1 (CO2CH3), 77.6 (C-1), 115.4 (C-3), 135.9 (C-4), 172.9 (CO2CH3), 212.3 (COMe); MS (70 eV) 213 (M++1, 1), 194 (2), 169 (76), 153 (10), 137 (35), 109 (100), 95 (13), 81 (13). Anal. Calcd for C11H16O4: C, 62.25; H, 7.59. Found: C, 62.50; H, 7.62.
3-Methoxy-3,6-dimethyl-4,5-dihydro-1[1H]-isobenzofuranone (28). A solution of 0.20 g (0.55 mmol) of 25 in dry THF (5 mL), under an N2 atmosphere and at 0 °C, was treated with 0.911 g (6.60 mmol) of anhydrous K2CO3 in dry MeOH (3.06 mL, 0.137 mol). After being stirred for 48 h at room temperature, EtOAc (75 mL) was added, and the mixture was washed with aqueous 5% HCl (3 × 20 mL) and with cold water until neutral. The organic layer was dried (Na2SO4), and the solvent was removed under vacuum. The residue was purified by column chromatography on silica gel (6 g, hexane/EtOAc, 9:1) to give 0.069 g (65%) of 28 as a pale yellow oil: Rf 0.85 (hexane/EtOAc, 7:3); IR (CH2Cl2) 1769, 1537, 1292, 1185, 1026 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.64 (s, 3H, CH3C-3), 1.92 (br s, 3H, CH3C-6), 2.40-2.56 (m, 4H, H-4, H-5), 3.21 (s, 3H, CH3O), 5.98-6.00 (m, 1H, H-7); 13C NMR (75 MHz, CDCl3) δ 19.9 (C-5), 22.8 (CH3), 23.1 (CH3), 28.1 (C-4) 50.9 (CH3O), 108.1 (C-3), 111.1 (C-7), 128.2 (C-6), 140.6 (C-7a), 154.2 (C-3a), 168.5 (C-1). Anal. Calcd for C11H14O3: C, 68.02; H, 7.26. Found: C, 67.87; H, 7.13.
Acknowledgments
We are indebted to Dr. Hugo A. Jiménez-Vázquez for reviewing the manuscript. We thank Fernando Labarrios for their help in spectrometric measurements. J.T. acknowledges DEPI/IPN (Grant 921769) and CONACYT (Grant 1570P) for financial support. A.O. is grateful to the Ludwig K. Hellweg Foundation for a scholarship.
References and Notes
1. (a) Waksman, S. A. The Actinomycetes, Ronald Press, New York, 1987. [ Links ] (b) Remers, W. A. The Chemistry of Antitumor Antibiotics, John Wiley & Sons, New York, 1979, Vol. 1, Chap. 2. [ Links ] (c) Crooke, S. T.; Du Vernay, V. H.; Mong, S. Molecular Pharmacology of Anthracyclines, in: Molecular Actions and Targets for Cancer Chemotherapeutic Agents, Chap 7, Academic Press, New York, 1981. [ Links ] (d) Myers, C. Cancer Chemother. 1986, 8, 52. [ Links ] (e) Yoshimoto, A.; Fujii, S.; Johdo, O.; Kubo, K.; Ishikura, T.; Naganawa, H.; Sawa, T.; Takeuchi, T.; Umezawa, H. J. Antibiot. 1986, 39, 902. [ Links ]
2. Arcamone, F. The Development of New Antitumor Anthracyclines, in: Medicinal Chemistry, Vol. 16, Chap. 1, Cassady, J. M.; Douros, J. D., Eds., Academic Press, New York, 1980. [ Links ]
3. Crooke, S. T. The Anthracyclines, in: Cancer & Chemotherapy, Vol. 3, Chap. 8, Academic Press, New York, 1981. [ Links ]
4. Arcamone, F. Doxorubicin, Anticancer Antibiotics, in: Medicinal Chemistry, Vol. 17, Academic Press, New York, 1981. [ Links ]
5. (a) Takiguchi, Y.; Mishima, H.; Okuda, M.; Terao, M.; Aoki, A.; Fukuda, R. J. Antibiot. 1980, 33, 1120. [ Links ] (b) Okazaki, T.; Ono, M.; Aoki, A.; Fukuda, R. J. Antibiot. 1983, 36, 438. [ Links ] (c) Mishima, H.; Ide, J.; Muramatsu, S.; Ono, M. J. Antibiot. 1983, 36, 980. [ Links ]
6. (a) Davies, H. G.; Green, R. H. Nat. Prod. Rep. 1986, 3, 87-121. [ Links ] (b) Davies, H. G.; Green, R. H. Chem. Soc. Rev. 1991, 20, 211-269. [ Links ] (c) Davies, H. G.; Green, R. H. Chem. Soc. Rev. 1991, 20, 271-339. [ Links ]
7. (a) Parmee, E. R.; Steel, P. G.; Thomas, E. J. J. Chem. Soc., Chem. Commun. 1989, 1250-1253. [ Links ] (b) Ley, S. V.; Anthony, N. J.; Armstrong, A.; Brasca, M. G.; Clarke, T.; Culshaw, D.; Greck, C.; Grice, P.; Jones, A. B.; Lygo, B.; Madin, A.; Sheppard, R. N.; Slawin, A. M. Z.; Williams, D. J. Tetrahedron 1989, 45, 7161-7194. [ Links ] (c) Karim, S.; Parmee, E. R.; Thomas, E. J. Tetrahedron Lett. 1991, 32, 2269-2272. [ Links ] (d) Hirama, M.; Noda, T.; Itô, S.; Kabuto, C. J. Org. Chem. 1988, 53, 706-708. [ Links ] (e) Danishefsky, S. J.; Armistead, D. M.; Wincott, F. E.; Selnick, H. G.; Hungate, R. J. Am. Chem. Soc. 1989, 111, 2967-2980. [ Links ]
8. (a) Reyes, A.; Aguilar, R.; Muñoz, A. H.; Zwick, J.-C.; Rubio, M.; Escobar, J.-L.; Soriano, M.; Toscano, R.; Tamariz, J. J. Org. Chem. 1990, 55, 1024-1034. [ Links ] (b) Aguilar, R.; Reyes, A.; Tamariz, J.; Birbaum, J.-L. Tetrahedron Lett. 1987, 28, 865-868. [ Links ]
9. (a) Andrade, R. M.; Muñoz, A. H.; Tamariz, J. Synth. Commun. 1992, 22, 1603-1609. [ Links ] (b) Orduña, A.; Zepeda, L. G.; Tamariz, J. Synthesis 1993, 375-377. [ Links ] (c) Ochoa, M. E.; Arias, M. S.; Aguilar, R.; Delgado, F.; Tamariz, J. Tetrahedron, 1999, 55, 14535-14546. [ Links ]
10. Kimball, S. D.; Walt, D. R.; Johnson, F. J. Am. Chem. Soc. 1981, 103, 1561-1563. [ Links ]
11. (a) Muzart, J. Chem. Rev. 1992, 92, 113-140. [ Links ] (b) Majetich, G.; Song, J.-S.; Ringold, C.; Nemeth, G. A.; Newton, M. G. J. Org. Chem. 1991, 56, 3973-3988. [ Links ] (c) Trachtenberg, E. N.; Carver, J. R. J. Org. Chem. 1970, 35, 1646-1653. [ Links ] (d) Trachtenberg, E. N.; Nelson, C. H.; Carver, J. R. J. Org. Chem. 1970, 35, 1653-1658. [ Links ]
12. For related methods, see: (a) Kende, A. S.; Tsay, Y.-g.; Mills, J. E. J. Am. Chem. Soc. 1976, 98, 1967-1969. [ Links ] (b) Smith, T. H.; Fujiwara, A. N.; Henry, D. W.; Lee, W. W. J. Am. Chem. Soc. 1976, 98, 1969-1971. [ Links ]
13. Broadhurst, M. J.; Hassall, C. H.; Thomas, G. J. Tetrahedron 1984, 40, 4649-4656. [ Links ]
14. (a) Blanchette, M. A.; Choy, W.; Davis, J. T.; Essenfeld, A. P.; Masamune, S.; Roush, W. R.; Sakai, T. Tetrahedron Lett. 1984, 25, 2183-2186. [ Links ] (b) Sundberg, R. J.; Bukowick, P. A.; Holcombe, F. O. J. Org. Chem. 1967, 32, 2938-2941. [ Links ]
15. (a) Etemad-Moghadam, G.; Seyden-Penne, J. Tetrahedron 1984, 40, 5153-5166. [ Links ] (b) Engler, T. A.; Falter, W. Synth. Commun. 1988, 18, 783-790. [ Links ] (c) Rodriguez, J.; Waegell, B. Synthesis 1988, 534-535. [ Links ]

