



![Estudio teórico de la reactividad química de 1,2,7-trimetil-6-(4- R-fenil)-1,6-dihidropirrolo[2,3-e]indoles frente a electrófilos](/img/en/prev.gif)






Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.44 n.1 Ciudad de México Jan./Mar. 2000
Investigación
Evaluación computacional del efecto anomérico absoluto
Gabriel Cuevas,*1 Julieta Tenorio2 y Fernando Cortés1
1 Instituto de Química, Universidad Nacional Autónoma de México, Circuito exterior, Ciudad Universitaria, Coyoacán 04510, México, D.F., México. E-mail: gecgb@servidor.unam.mx
2 Escuela de Ciencias Químicas, Universidad La Salle, Benjamín Franklin 47, Hipódromo Condesa, 06140 México, D.F., México.
Recibido el 3 de febrero del 2000.
Aceptado el 7 de marzo del 2000.
Resumen
Se propone la determinación del efecto anomérico absoluto, como una alternativa computacional para evaluar el exceso en la preferencia por la posición axial que muestran los sustituyentes electronegativos sitos en la posición α al heteroátomo anular de un compuesto heterocíclico (posición anomérica), respecto a la preferencia que muestran cuando son sustituyentes en el ciclohexano. El efecto anomérico absoluto indica la diferencia en la preferencia que por la posición axial muestra un sustituyente electronegativo en presencia y ausencia de deslocalización electrónica, manteniéndose la misma geometría molecular. Esto se logra restando a la energía total, la energía de la molécula hipotética de Lewis (en donde todos los electrones se encuentran estrictamente localizados en enlaces o en pares electrónicos no compartidos) de los confórmeros implicados en el equilibrio correspondiente. Aplicando esta metodología a nivel Becke3LYP/6-31G(d,p) se encuentra que el efecto anomérico que muestran el cloro en el 1,3-dioxano y el flúor, cloro, y los grupos SMe, +PH3, y CO2Me en el 1,3-ditiano son de naturaleza estereoelectrónica, mientras que la preferencia del flúor, OMe, NH2 en el 1,3-dioxano y el grupo P(O)Me2 en el 1,3-ditiano no lo son, por lo que su comportamiento conformacional tiene un origen no estereoelectrónico. También se muestra que los efectos anoméricos que no tienen un origen estereoelectrónico pueden modificar la geometría molecular de acuerdo con el patrón requerido por el modelo de doble enlace-no enlace, como recientemente se ha sido propuesto por Perrin.
Palabras clave: Efecto anomérico absoluto, análisis conformacional, química computacional, orbitales naturales de enlace, análisis de población.
Abstract
The Absolute Anomeric Effect (AAE, Eq. 3), is proposed as a computational alternative in the evaluation of the excess in the axial preference shown by electronegative substituents located at the α position to the anular heteroatom of an heterocyclic compound (anomeric position), in both the presence and the absence of electron ic delocalization, retaining the same molecular geometry. The aim of the determination of the anomeric effect is computationally accessi ble by applying the natural bond orbital analysis (NBO), in order to compare the energy of the hypothetical molecules lacking electronic delocalization (Lewis molecules, in which the electrons are strictly located in bonds and lone pairs) with the fully delocalized molecules retaining the same geometry, and to evaluate the anomeric effect in terms of equation 3. The role of the Lewis molecules is the same as when cyclohexane is used experimentally, with the advantage that they are really stereoelectronically inert. By applying this methology to cyclic molecules at B3LYP/6-31G(d,p) and HF/6-31G(d,p)// B3LYP/6-31G(d,p) levels of theory, we found out that the anomeric effect shown by Cl in 1,3-dioxane; F, Cl, SMe, PH3 and CO2Me groups in 1,3-dithiane is of stereoelectronic nature, while the preference of F, OMe and NH2 in 1,3-dioxane and P(O)Me2 group in 1,3-dithiane is not; besides, this methology shows that anomeric effects with no stereoelectronic origin can modify the molecular geometry, in agreement with the geometric pattern required by double bond-non bond model, as has been recently proposed by Perrin.
Key words: Anomeric effect, absolute anomeric effect, conformat ional analysis, computational chemistry, natural bond orbitals, population analysis.
Dedicado a la memoria del Dr. Raúl Cetina Rosado
Introducción
El efecto anomérico se define como el exceso en la preferencia por la posición axial que muestran los sustituyentes electronegativos sitos en la posición α al heteroátomo anular (posición anomérica), respecto a la preferencia que muestra el mismo sustituyente en el ciclohexano, y que se supone es desfavorecida en términos estéricos debido a la repulsión que sufre el sustituyente axial de la posición C2, con los grupos de las posiciones 4,6-syn-diaxiales (Fig. 1)[1].

Los primeros intentos por determinar la magnitud del efecto anomérico se deben a Eliel et al. [2] y a Anderson y Sepp,[3] y se determina de acuerdo con la ecuación 1.
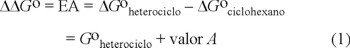
En donde ΔGoheterociclo corresponde a la energía libre con formacional del sustituyente en el sistema heterocíclico en estudio y ΔGociclohexano corresponde a la del ciclohexano, a cuyo valor negativo se denomina también valor A y que se considera un índice de las interacciones estéricas [4].
En esta aproximación se asume: (1) que las interacciones estéricas que sufre el sustituyente de la posición anomérica permanecen constantes al pasar del ciclohexano al sistema heterocíclico, (2) que el ciclohexano es estereoelectrónicamente inerte, lo que permite concebir la idea de aislar la energía proveniente de la deslocalización electrónica, y (3) que se suprime cualquier interacción electrostática posible, favorecida por la introducción de átomos electronegativos en el sistema anular.
Esta manera de medir el efecto anomérico ha generado serias controversias; por ejemplo, con base en la ecuación 1 se determinó erróneamente que el 2-carbometoxi-tetrahidropirano presenta un efecto anomérico inverso (un exceso en preferencia por la posición ecuatorial) [5]. El origen de estos problemas se debe a que la introducción de los heteroátomos en el sistema anular ocasiona cambios profundos en la geometría molecular [6] (cambio en las longitudes de enlace, en los ángulos de valencia y en los ángulos diedros) y por lo tanto, cambios en el ambiente estérico que experimenta el sustituyente en evaluación.
Con la intención de corregir los cambios en la geometría molecular, Franck [7] correlacionó linealmente la energía libre conformacional de una serie de sustituyentes "electrónicamente inertes" (los grupos metilo, etilo, iso-propilo y ter-butilo) en el ciclohexano, con el ΔGo de ese sustituyente en el tetrahidropirano. Así se encontró que la apariencia estérica del grupo metilo se incrementa 1.53 veces al ser transferido del ciclohexano al tetrahidropirano. Con esta aproximación se demostró que los grupos metilamino y dimetilamino muestran efectos anoméricos normales [7]. El grupo ter-butilo tiene un valor A de 4.9 kcal/mol, mientras que cuando es sustituyente en el 1,3-ditiano, el ΔGo es de 2.72 kcal/mol. La introducción de este factor de escalamiento permitió corregir el valor del efecto anomérico encontrado para el 2-difenilfosfinoíl-1,3-ditiano de 3.74 kcal/mol a 2.64 kcal/mol al aplicar la ecuación 2 [8].

en donde α corresponde al valor de 0.6 (2.72 /4.9) para este caso y permite estimar el valor de ΔΔHo (efecto anomérico entálpico) de 3.39 kcal/mol efectuando la corrección sobre el ΔHo, como se ha sugerido por Booth, para eliminar contribuciones entrópicas en la evaluación de las preferencias conformacionales [9].
Sin embargo, el intercambio de grupos "inertes" de ciclohexano a otros sistemas heterocíclicos no es lineal [7], por lo que no puede ser generalizado y tampoco puede ser aplicado a la evaluación de efectos anoméricos en sistemas no cíclicos, en donde el efecto anomérico se manifiesta por la preferencia por el confórmero gauche respecto al anti. De tal forma, nuevamente aparecen problemas de interpretación, aunque desde el punto de vista experimental, parece no haber alguna alternativa mejor. Sin embargo, debe tomarse en cuenta que la incorporación de los heteroátomos ocasiona cambios electrónicos, estereoelectrónicos y electrostáticos, por lo que las contribuciones estereoelectrónicas pueden no corresponder a la diferencia indicada en la ecuación 2, que es la razón del empleo del ciclohexano como referencia en las ecuaciones 1 y 2 [2].
Desde el punto de vista computacional [10], el efecto anomérico se ha definido como la diferencia positiva entre la energía del confórmero axial (o gauche) y el ecuatorial (o anti-periplanar). Esta manera de medirlo, aunque simple, no mantiene el espíritu original de la definición en términos de un exceso por las preferencias axial o gauche. También se han efectuado cálculos del valor A mediante la introducción de potenciales estéricos átomo-átomo, [11,12] determinados tanto con mecánicas moleculares [13,14] como por métodos semiempíricos y ab initioaplicados al estudio de este fenómeno [15].
El análisis empleando orbitales naturales de enlace (ONE) permite una aproximación única a la evaluación del efecto anomérico desde el punto de vista computacional[16]. El aná lisis ONE ha sido ampliamente utilizado para estudiar tanto funciones de onda como densidades electrónicas obtenidas mediante cálculos ab initio [17], empleando tanto la aproxi mación de Hartree-Fock como la de Kohn-Sham y sus funda mentos han sido ampliamente discutidos [16]. Este análisis permite separar la energía molecular en dos contribuciones fundamentales, la energía total (donde la deslocalización está presente) y la energía de la molécula de Lewis (ELew ) correspondiente a la que tendría la molécula hipotética de Lewis en donde la hiperconjugación está ausente, y en la que los elec trones se encuentran estrictamente localizados en enlaces y en pares electrónicos no compartidos. Esto es posible ya que la matriz de Fock en la base ONE es diagonal y los elementos fuera de la diagonal corresponden a las interacciones estereoe lectrónicas [18]. Haciendo cero estos elementos de matriz y efectuando un cálculo en un campo autoconsistente se obtiene la energía de la molécula hipotética. Esta información es útil para conocer la energía de deslocalización: Edeloc = Etotal − ELew. Las propiedades de la matriz de Fock en términos de or bitales naturales se han discutido y revisado ampliamente con anterioridad [19].
El análisis ONE permite estimar el efecto anomérico en el sentido de Eliel (Eq. 1), pues es posible conocer la energía de la molécula con la misma geometría pero en ausencia de deslocalización electrónica, considerando sólo interacciones estéricas y electrostáticas por medio de la ELew. El efecto anomérico absoluto se puede calcular entonces, de acuerdo con la ecuación 3:

El cálculo del Efecto Anomérico Absoluto permite mantener la geometría molecular, eliminar por completo la deslocalización electrónica y mantener las interacciones electrostáticas presentes en la molécula. De esta forma se satisface completamente el deseo original de considerar al efecto anomérico como un índice de la deslocalización electrónica, aunque debe considerarse que paralelamente las cargas puntuales se deben incrementar y las interacciones electrostáticas se pueden potenciar y sobreestimar.
En este trabajo se muestran los resultados de evaluar el efecto anomérico absoluto en los sistemas heterocíclicos que se muestran en la Fig. 1 empleando el programa Gaussian 92/DFT [20] y el programa NBO 4.0 [21].
Resultados y discusión
La primera racionalización del efecto anomérico fué dada por Edward [22] en términos electrostáticos, pero con la finalidad de explicar los patrones geométricos experimentales se propuso un segundo modelo que se apoya en la deslocalización de los pares electrónicos no compartidos del heteroátomo anular (Y) en el orbital de antienlace C-X [23,24] (Fig. 2). Esto en términos de Cieplak [25] corresponde a una interacción de tipo nY → σ*C-X. En términos de la Teoría de Enlace-Valencia este efecto se puede representar mediante un híbrido de doble enlace-no enlace [1]. La participación de esta interacción en el confórmero axial repercute en el elongamiento del enlace C-X, en el acortamiento del enlace C-Y, el aumento del ángulo Y-C-Y y en la disminución del ángulo Y-C-Y-C [23]. Recientemente, Perrin et al. [26,27] describieron que el acortamiento del enlace C-Y puede ser explicado satisfactoriamente en términos de interacciones puramente electrostáticas, por lo que la modificación de la geometría no es una condición necesaria y suficiente para que se deba invocar al modelo sugerido por Altona et al. [24].

En la Tabla 1 se puede observar que el patrón de acortamiento y elongación de enlaces es satisfecho por la mayor parte de los equilibrios estudiados excepto en 11 y 13. En el equilibrio 11 no hay diferencias importantes entre ambos confórmeros, mientras que el equilibrio 13 es el único sistema en donde el patrón se rompe completamente, tal y como se describió experimentalmente [1, 4, 8, 28].
La energía de estos sistemas a nivel Becke3LYP/6-31G (d,p) se incluye en la tabla 2. Como consecuencia del efecto anomérico, se espera que el confórmero axial sea más estable que el ecuatorial. Se acepta que la preferencia que los sustituyentes muestran por adoptar la posición ecuatorial en el ciclohexano se debe a efectos netamente estéricos, aunque el ciclo hexano no es estereoelectrónicamente inerte, ya que a nivel Becke3LYP/6-31G(d,p) tiene una energía total de -235.89717 mientras que su correspondiente molécula de Lewis tiene -235.59751 hartrees, lo que genera una diferencia de 188.03 kcal/mol de energía de deslocalización, equivalente al 0.13% de la energía total. A nivel HF/6-31G(d,p) estas energías son -234.22537 y -233.89234 Hartrees, por lo que la energía de deslocalización es de 208.98 kcal/mol, equivalente a un 0.14%. Este hecho se encuentra apoyado experimentalmente mediante las constantes de acoplamiento 1JC-H ya que el ciclohexano muestra un efecto Perlin normal [29] y en el corri miento químico a campo alto de los hidrógenos axiales en el espectro de resonancia magnética nuclear de protón [30]. Por esto es de esperar que si se elimina la deslocalización electró nica en los sistemas heterocíclicos en estudio, dominará el efecto estérico y por lo tanto se observará la preferencia por el confórmero ecuatorial.

De la manera en que están descritos los equilibrios con formacionales (Fig. 1), si se obtiene una diferencia de energía total positiva y una diferencia de energía de Lewis negativa y menor en términos absolutos, se obtiene un efecto anomérico absoluto positivo originado por la participación dominante de efectos estereoelectrónicos. Este fenómeno puede esperarse, ya que el confórmero axial es favorecido cuando la deslocali zación electrónica domina, mientras que se favorece el ecuatorial cuando se manifiesta el efecto estérico.
Cuando la energía de Lewis es positiva indica que el sus tituyente prefiere la posición axial en ausencia de deslocalización electrónica, e implica que la interacción nY → σ*C-X no participa en la estabilización. Si el valor absoluto de la energía total es menor, se encuentra un EAA negativo (Ec. 3), indica tivo de un efecto anomérico de origen posiblemente electros tático, de igual manera al caso en el que en presencia de deslo calización electrónica hay preferencia por la posición ecuatorial (valor negativo de ΔEtotal) pero preferencia axial en la mo lécula hipotética de Lewis. Finalmente, si ambas energías son negativas, la preferencia por la posicion ecuatorial tendrá una componente estereoelectrónica.
Como se observa en la Tabla 2, en el 1,3-dioxano, cuando en la posición anomérica se localizan el átomo Cl y en el 1,3-ditiano cuando los sustituyentes son los grupos Cl, F, SMe, PMe2, PH3, COMe presentan un efecto anomérico de origen estereolectrónico.
El cálculo predice que para el equilibrio 1, 1-ax es favorecido en presencia de deslocalización electrónica y que 1-eq es el favorecido en ausencia de interacciones estereoelectrónicas. Comportamiento donde las interacciones descritas dominan al efecto estérico, y que corresponde con lo esperado para un efecto anomérico de origen estereoelectrónico.
Cuando se revisan los dioxanos con F, OMe, NH2 y O(-) y el ditiano con P(O)Me2 se encuentra una diferencia negativa que corresponde a un efecto anomérico en el que los factores no estereoelectrónicos son responsables del comportamiento conformacional. La preferencia del átomo de O(-) en el 1,3-dioxano es por la posición axial (Tabla 2). A un nivel mayor de teoría, [Becke3LYP/6-311+G(d,p)//Becke3LYP/6-31G (d,p)] la diferencia en energía total entre el confórmero axial y el ecuatorial de este compuesto es de 2.03 kcal/mol en favor del confórmero axial. El EAA que presenta 7 implica un origen no estereoelectrónico, congruente con el hecho de que la interacción nO → σ*C-O(-) no es posible porque la diferencia en la energía de los orbitales que interactuan es muy grande, además del exceso de carga sobre el átomo de oxígeno en el híbrido involucrado, aunque el patrón geométrico si corres ponde a este modelo, dando evidencia, como ha sido demostrado por Perrin et al., [26,27] de que los efectos electrostáti cos pueden modificar la geometría molecular de la misma forma. El modelo de Edward [22] permite justificar esta preferencia conformacional.
También es posible que una interacción de tipo electrostático entre algunos de los hidrógenos de las posiciones 4,6-syn diaxiales y el sustituyente en C2 pudieran ser el origen de esta preferencia. Dada la naturaleza de esta interacción, la distancia entre los centros que interactúan electrostáticamente es muy importante. En la Tabla 1 se incluye la distancia X-Hax4,6. Se puede observar que esta distancia oscila entre 2.54 y 2.62 Å para el caso de los derivados del 1,3-dioxano y de 3.146 para el ditiano 13-ax. Sin embargo, en este caso la interacción no es con el átomo directamente unido al carbono anomérico, sino con el átomo de oxígeno ubicado a 2.489 Å, distancia a las que se puede esperar una interacción entre los dos átomos involucrados en términos de la suma de sus radios de van der Waals.
Llama entonces la atención el caso del equilibrio 14, en donde el átomo de oxígeno del carbonilo podría mostrar un comportamiento similar al del fosfinoílo. En el caso de 14-ax el carbonilo no apunta hacia el centro del anillo, sino hacia uno de los átomos de hidrógeno axiales, como sucede en 13-ax. Experimentalmente, [31] se ha determinado que la capacidad para formar puentes de hidrógeno de los óxidos de fosfina es mucho mayor a la de las cetonas.
En el caso del dioxano con OH se observa que bajo el efecto estereoelectrónico prefiere la posición ecuatorial, mientras que en ausencia de este efecto la posición axial es la adoptada produciendo una diferencia negativa, indicativa de un efecto anomérico de tipo exo.
En el caso de 5 ax un átomo de hidrógeno unido al de ni trógeno ocasiona una interacción repulsiva ya que se encuentra a 2.714 Å de los protones de las posiciones 4,6-syn diaxiales. Este hecho puede estar estrechamente vinculado con el llamado efecto anomérico inverso, que experimentan este tipo de moléculas, que tiene una pequeña componente estereoelectró nica y una fuerte preferencia por el confórmero ecuatorial de origen no estereoelectrónico, que incluye interacciones estéricas y electrostáticas, como lo han propuesto Perrin et al. [1c, 32].
Es importante destacar que la magnitud relativa del efecto anomérico absoluto para los sistemas cuyo origen es estereoe lectrónico, sigue la tendencia esperada por el incremento de la electronegatividad del sustituyente, como se ve en la tendencia de los equilibrios 8, 10, 11, 12.
En la Tabla 3 se muestran las energías obtenidas de la op timización completa de la geometría de las moléculas involucradas en los equilibrios 2 a 4, 9 y 13 a nivel HF/6-31G(d,p), confirmándose el origen no estereoelectrónico de la preferen cia por el confórmero axial de estos compuestos, discutidos anteriormente en el marco de la Teoría de Funcionales de la Densidad.

Como cualquier análisis de población en el espacio de Hilbert, [33] el aquí empleado es sensible al nivel de teoría con el que se calcula. Al contrastar los resultados de las Tablas 1 y 3 se encuentra una diferencia sustancial entre la energía de Lewis del equilibrio 13. Esto se puede deber a una sobre estimación de las interacciones electrostáticas debidas a la localización electrónica. Otros equilibrios en donde esta energía parece sobre estimada a nivel Becke3LYP/6-31G(d,p) son el 5 y el 7, en donde el efecto electrostático puede tener una participación considerable.
Conclusiones
El efecto anomérico absoluto (EAA, Ec. 3) obtenido mediante la determinación de la energía de las moléculas hipotéticas de Lewis calculado con el programa NBO, [19,21] permite determinar el efecto de la deslocalización electrónica en el equilibrio conformacional. Con este análisis es posible diferenciar los efectos anoméricos de origen estereoelectrónico de aquéllos que no tienen este origen.
Así, la determinación del efecto anomérico absoluto (que incluye sólo interacciones estereoelectrónicas) difiere del que se obtiene por comparación con el ciclohexano, porque en este se disminuyen las interacciones estereoelectrónicas y se suprimen completamente las interacciones electrostáticas generadas por la presencia de átomos electronegativos.
El concepto de efecto anomérico, considerado como un exceso de la preferencia por la conformación axial parece no aplicable a sistemas que, en ausencia de interacciones electrostáticas muestran la misma preferencia; sin embargo es explicable, pues el efecto anomérico puede tener un origen distinto al estereoelectrónico.
Métodos computacionales
La optimización completa de la geometría (sin restricciones por consideraciones simétricas) de todas las moléculas analizadas en este trabajo se efectuó en el marco de la Teoría de Funcionales de la Densidad usando el funcional híbrido Becke3LYP y la base doble ζ de valencia dividida 6-31G(d,p) que incluye polarización en los átomos pesados y en los ligeros, misma base que se empleó en los cálculos HF utilizando el programa G92 [20].
Como es bien sabido en Becke3LYP, el funcional de intercambio se combina con un funcional de correlación local y de gradiente corregido. El funcional de correlación usado es el C*ECLYP + (1-C)*ECVWN, en donde LYP es el de correlación de Lee, Yang y Parr, [34] que incluye por su parte, términos locales y de gradiente corregido y el término VWN es el funcional de Vosko, Wilk and Nusair de 1980 que ajusta la solución del gas uniforme, denominada correlación de densidad local de spin [35]. VWN permite proveer el exceso de correlación local requerida, ya que LYP contiene un término local equivalente a VWN [36].
El análisis de orbitales naturales se efectuó con la versión 4 del programa NBO incluida en G92 [21]. La energía de las moléculas hipotéticas de Lewis se obtuvo adicionando la palabra NOSTAR en la ruta de cálculo del programa NBO.
Agradecimientos
Este trabajo se realizó con el apoyo a los proyectos 32420-E del Consejo Nacional de Ciencia y Tecnología (CONACyT) e IN-107597 de la Dirección General de Apoyo del Personal Académico (DGAPA) de la Universidad Nacional Autónoma de México. También agradecemos a la Dirección General de Servicios de Cómputo Académico (DGSCA) por permitirnos el uso de la supercomputadora CRAY-YMP4/64 y al Departamento de Sistemas Académicos de la Universidad la Salle por las facilidades brindadas. Julieta Tenorio y Fernando Cortés también agradecen las becas otorgadas por el CONACyT.
Referencias
1. a) Kirby, A.J. The anomeric effect and Related Stereoelectronic Effects at Oxygen; Springer Verlag: Berlin, 1983 p.100-102. [ Links ] b) Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry; Pergamon Press: New York, 1983. [ Links ] c) Juaristi, E.; Cuevas, G. The Anomeric Effect. CRC Press: Boca Raton, FL. 1994. [ Links ]
2. Eliel, E.L.; Allinger, N.L.; Angyal, S.J.; Morrison,G.A.; Conformational Analysis Wiley, New York, 1967. p. 377. [ Links ]
3. Anderson, C.B.; Sepp, D.T. Tetrahedron 1968, 24, 1707-1716. [ Links ]
4. Juaristi, E. Acc. Chem. Res. 1989, 22, 357-364. [ Links ]
5. Anderson, C.B.; Sepp, D.T. J. Org. Chem. 1968, 33, 3272-3276. [ Links ]
6. Eliel, E.L.; Giza, C.A. J. Org. Chem. 1968, 33, 3754-3758. [ Links ]
7. Franck, R.W. Tetrahedron 1983, 39, 3251-3252. [ Links ]
8. Juaristi, E.; Valle, L.; Mora-Uzeta, C.; Valenzuela, B.A.; Joseph-Nathan, P.; Fredrich, M.F. J. Org. Chem. 1982, 47, 5038-5039. [ Links ]
9. Booth, H.; Grindley, T.B., Khedhair, K.A. J. Chem. Soc. Chem. Commun. 1982, 1047-1048. [ Links ]
10. Graczyk, P.P.; Mikolajczyk M. The Anomeric Effect: Origin and Consequences in Topics in Stereochemistry Eliel, E.L.; Wilen, S.H. (Eds.), 1994, 21, p. 166. [ Links ]
11. Vijayalakshmi, K.S.; Rao, V.S.R. Carbohyd. Res. 1972, 22, 413-424. [ Links ]
12. Abe, A. J. Am. Chem. Soc. 1976, 98, 6477-6480. [ Links ]
13. Allinger, N.L. J. Am. Chem. Soc. 1977, 99, 8127-8134. [ Links ]
14. Woods, R.J.; Andrews, C.W.; Bowen, J.P. J. Am. Chem. Soc. 1992, 114, 850-858. [ Links ]
15. Cálculos semiempíricos: a) Tvaroska, T.; Bleha, T. En: Advances in Carbohydrate Chemistry and Biochemistry, Tipson, R.S.; Holton, D. (Eds).; Academic Press: San Diego, 1989; pp. 75-81 y referencias ahí incluidas. [ Links ] b) Tvaroska, I.; Carver, J.P. J. Chem. Res. (S) 1991, 6. [ Links ] c) Woods, R.J.; Szarek, W.A.; Smith, Jr., V.H. J. Chem. Soc., Chem. Commun. 1991, 334-337. [ Links ] Cálculos ab initio: d) Wolfe, S.; Pinto, B.M.; Varma, V.; Leung, R.Y.N. Can. J. Chem. 1990, 68, 1051-1062. [ Links ] e) Schleyer, P.v.R.; Jemmis, E.D.; Spitznagel, G.W. J. Am. Chem. Soc. 1985, 107, 6393-6396. [ Links ] f) Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. J. Am. Chem. Soc. 1982, 104, 2797-2803. [ Links ]
16. a) Reed, E.A.; Weinstock, R.B.; Weinhold, F.J. J. Chem. Phys. 1985, 83, 735-746. [ Links ] b) Reed, E.A.; Weinhold, F. J. Chem. Phys. 1985, 83, 1736-1740. [ Links ] d) Reed, E.A.; Curtiss, L.A.; Weinhold, F. Chem. Rev. 1988, 88, 899-926. [ Links ]
17. Petillo, P.A.; Lerner, L.E. en The Anomeric Effect and Associated Stereoelectronic Effects. ACS Thatcher, G.R. R. (Ed.) Symposium Series 539, Washington: 1993. p. 156. [ Links ]
18. Salzner, U.; Schleyer, P.v.R. J. Org. Chem. 1994, 59, 2138-2155. [ Links ]
19. Salzner, U.; Schleyer, P.v.R. J. Am. Chem. Soc. 1993, 115, 10231-10236. [ Links ]
20. Gaussian 92/DFT, Revision G.2. Frisch,M. J.; Trucks,G. W.; Schlegel,H. B.; Gill, P. M. W.; Johnson, B.G.; Wong, M. W.; Foresman, J. B.; Robb, M. A.; Head-Gordon, M.; Replogle, E. S.; Gomperts, R.; Andres, J. L.; Raghavachari, K.; Binkley, J. S.; Gonzalez, C.; Martin, R. L.; Fox, D. J.; Defrees, D. J.; Baker, J.; Stewart, J. J. P.; Pople, J. A. Gaussian, Inc., Pittsburgh PA, 1993. [ Links ]
21. NBO 4.0. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1994. [ Links ]
22. Edward, J.T. Chem. and Ind. 1955, 1102-1104. [ Links ] 48 Rev. Soc. Quím. Méx. Vol. 44, Núm. 1 (2000) Gabriel Cuevas et al.
23. Brockway, L.O. J. Phys. Chem. 1937, 41, 185-195. [ Links ]
24. Romers, C.; Altona, C.; Buys, H.R.; Havinga, E. Top. Stereochem. 1969, 4, 39. [ Links ] de Wolf, N.; Romers, C.; Altona, C. Acta Crystallogr. 1967, 22, 715-719. [ Links ]
25. a) Cieplak, A.S. J. Am. Chem. Soc. 1981, 103, 4540-4552. [ Links ] b) Cieplak, A.S.; Tait, B.D.; Johnson, C.R. J. Am. Chem. Soc. 1989, 111, 8447-8462. [ Links ]
26. Perrin, C.L. en The Anomeric Effect and Associated Stereoelectronic Effects. ACS Thatcher, G.R. R. (Ed.) Symposium Series 539, Washington: 1993. p. 70-96 [ Links ]
27. Perrin, C.L.; Armstrong, K.B.; Fabian, M.A. J. Am. Chem. Soc. 1994, 116, 715-722. [ Links ]
28. a) Juaristi, E.; Valle, L.; Valenzuela, B.A,: Aguilar, M.A. J. Am. Chem. Soc. 1986, 108, 2000-2005, [ Links ] b) Juaristi, E.; Cuevas, G. J. Am. Chem. Soc. 1993, 115, 1313-1316. [ Links ]
29. Juaristi, E.; Cuevas, G.; Vela, A. J. Am. Chem. Soc. 1994, 116, 5796-5804. [ Links ]
30. Cuevas, G.; Juaristi, E.; Vela, A. J. Mol. Struct. (Theochem) 1997, 418, 231-241. [ Links ]
31. Taylor, R.; Kennard, O. J. Am. Chem. Soc. 1982, 104, 5063-5070. [ Links ]
32. Perrin, C.L.; Armstrong, K.B. J. Am. Chem. Soc. 1993, 115, 6825-6834. [ Links ]
33. Bachrach, S.M. Population analysis and electron densities from Quantum Mechanics. en Reviews in Computational Chemistry. Lipkowitz, K.B.; Boyd, D.B. (Eds.) VCH Publishers, Inc. New York, 1994, p. 171. [ Links ]
34. a) Lee, C.; Yang, W.; Parr, R.G. Phys. Rev. 1988, B37, 785-789. [ Links ] b) Miehlich, B.; Savin, A. Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200-206. [ Links ]
35. Vosko, S.H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200-1211. [ Links ]
36. Becke, A.D. J. Chem. Phys. 1988, 88, 1053-1062. [ Links ] Becke, A.D. Phys. Rev. A. 1988, 38, 3098-3100. [ Links ]

