




![Estudio teórico de la reactividad química de 1,2,7-trimetil-6-(4- R-fenil)-1,6-dihidropirrolo[2,3-e]indoles frente a electrófilos](/img/es/next.gif)





Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.44 no.1 Ciudad de México ene./mar. 2000
Investigación
Estudio ab initio de la estructura electrónica y conformacional de oligómeros de tiofeno
Angélica Raya y Marco Antonio Mora*
Departamento de Química, Universidad Autónoma Metropolitana, Campus Iztapalapa, Apdo. Postal 55-534, México, D.F., CP 09340, Tel. (+52) 5804 4675, Fax (+52) 5804 4666. E-mail: mam@xanum.uam.mx
Recibido el 21 de enero del 2000.
Aceptado el 1 de marzo del 2000.
Resumen
En este trabajo se presentan los resultados obtenidos en el estudio ab-initio, a nivel RHF/6-31G(d), de la estructura electrónica y conformacional de oligómeros de tiofeno (n = 2 − 8). Para cada oligómero se determinó la geometría de mínima energía, la estructura electrónica, el potencial de ionización vertical y la energía de excitación. Debido a la longitud de los enlaces C - C (1.462 Å) y C = C (1.352 Å), la estructura de los oligómeros se caracterizó como tipo bencenoide. Los resultados obtenidos permiten proponer como modelo representativo del comportamiento de los oligotiofenos al oligómero con 6 anillos.
Palabras clave: Oligotiofenos, polímeros conjugados, conductores poliméricos.
Abstract
In this work we discuss from a theoretical point of view the relationship between the electronic structure and the polythiophene backbone chemistry. We have obtained ab-initio geometry and electronic structure for oligomers of thiophene (n = 2 − 8) using a 6-31G(d) basis set at the Hartree-Fock-SCF level of theory. We have also calculated vertical ionization potentials and excitation energies. The oligomers shows a benzenoid structure, which was characterized by the C - C and C = C bond lengths with values of 1.462 and 1.352 Å respectively. The results obtained in our calculations allow us propose an oligomer with 6 rings as a representative model of oligothiophenes behaviour.
Keywords: Oligothiophenes, conjugated polymers, polymeric conductors.
Dedicado a la memoria del Dr. Raúl Cetina
Introducción
Por su variedad de aplicaciones en electrónica y fotoelectrónica, los oligómeros y polímeros de tiofeno α-conjugados se han convertido en materiales atractivos para el desarrollo e implementación de nuevas tecnologías. Los politiofenos son materiales amorfos y no homogéneos, mientras que los oligotiofenos son compuestos bien definidos estructuralmente y fácilmente procesables, por lo que constituyen modelos cuyo estudio permitirá comprender las peculiaridades estructurales y electrónicas que controlan el transporte de carga y procesos ópticos observados en politiofenos.
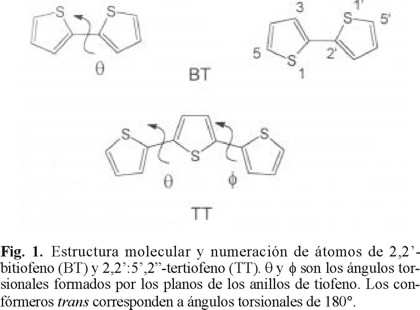
Una explicación de las propiedades ópticas y del fenómeno de conducción en materiales no metálicos se basa en el modelo teórico que considera que los polímeros conductores poseen una estructura con conjugación de simples y dobles ligaduras a lo largo de la cadena polimérica. Esta deslocalización intramolecular de electrones ϖ depende del traslape entre orbitales pz de los átomos de carbono α de anillos adyacentes (Fig. 1) y es gobernada por la rotación interna alrededor de los enlaces interanulares. El grado de planaridad de la estructura determina la extensión de la conjugación efectiva y el ancho de las bandas ϖ, incluyendo al band-gap óptico [1]. La flexibilidad de la cadena disminuye la conjugación electrónica, un factor que ha sido señalado como el responsable de los cambios de color observados en politiofenos sustituidos que muestran termocromismo [2, 3].
Los cálculos de estructura electrónica permiten predecir diversas propiedades moleculares, como geometrías de equili brio, barreras de torsión y energías de excitación electrónica de sistemas atómicos y moleculares [4]. Durante mucho tiem po, la magnitud de los sistemas poliméricos restringió su estu dio teórico a cálculos a nivel semiempírico o a aquéllos que solamente consideran a los electrones de valencia. El vertigi noso avance logrado en materia de cómputo (hardware y soft ware) durante la última década del siglo XX ha permitido es tudiarlos detalladamente con diversas metodologías y determi nar la relación existente entre su estructura electrónica y algunas de sus propiedades fisicoquímicas.
La literatura registra resultados teóricos interesantes en la descripción de la geometría y estructura electrónica de oligo tiofenos. Cálculos a nivel semiempírico MNDO predijeron es tructuras totalmente planares, pero es sabido que en solución ocurre torsión de la cadena [5]. A nivel ab initio, con el esquema Hartree-Fock [1, 6] se determinaron razonablemente las geometrías de equilibrio; al incorporar efectos de correlación electrónica con MP2 [1, 7] se obtuvo una excelente descrip ción de aspectos energéticos al realizar análisis conformacional. En el caso particular de 2,2'-bitiofeno (BT), tanto HF como MP2 predijeron apropiadamente la existencia de mínimos gauche y los valores para las barreras rotacionales [1]. De acuerdo con los resultados obtenidos en el análisis conformacional de la molécula de BT y sus respectivos cationes y dicationes, Alemán y Julià [7] señalan que el nivel de teoría mínimo necesario para realizar estudios de esta naturaleza es HF/6-31G(d).
Orti y col. [8] incorporaron efectos de correlación electrónica utilizando el contexto de la Teoría de Funcionales de la Densidad y determinaron que esta metodología falla al describir la energética de la rotación interna de BT, debido a que favorece las conformaciones planares sobre las perpendiculares y predice valores hasta dos veces mayores que los obtenidos con MP2 para las barreras de interconversión entre los confórmeros s-cis y s-trans. Al utilizar funcionales locales, el mínimo gauche desaparece completamente y dichas barreras rotacionales tienen un valor hasta tres veces mayor que el obtenido con MP2.
Utilizar MP2 para estudiar oligómeros con más de cuatro anillos de tiofeno requeriría de un gasto computacional considerable. Degli y Zerbetto [6] estudiaron el espectro vibracional del hexatiofeno y encontraron que en el caso de los parámetros geométricos de interés, C - C, C = C y C2' - C2 - C3 (Fig. 1), HF/6-31G(d) predice valores más próximos a resultados experimentales que los obtenidos con los funcionales BLYP y B3LYP. Brédas ha aplicado el Hamiltoniano de Valencia Efectiva (VEH) [9] al campo de los polímeros conductores, obteniendo para gaps valores muy cercanos a los valores experimentales.
El impacto de estudios de esta naturaleza radica en que un entendimiento razonado de las propiedades y de los procesos a nivel molecular debe incrementar nuestro conocimiento en tal magnitud que sería legítimo esperar que en un tiempo razonablemente corto, el desarrollo guiado de nuevos materiales poliméricos, con mejores características y nuevas aplicaciones, fuera posible.
Con el objeto de evaluar el impacto de la torsión de la ca dena sobre las propiedades electrónicas del politiofeno y ca racterizar el tipo de estructura predominante (bencenoide o quinoide), en este trabajo se describe la geometría y la estructura electrónica de diversos oligómeros de tiofeno (n = 2 - 8 unidades), utilizando métodos mecánico cuánticos ab initio, a nivel HF/6-31G(d). Un grado de libertad geométrico relevante en los oligómeros es la rotación alrededor del enlace interanu lar C - C, por lo que se realizó un análisis detallado del poten cial torsional para el dímero (BT) y el trímero, 2,2':5',2"-tertiofeno (TT) y se analizó la conformación adoptada por las di ferentes cadenas en términos del comportamiento de las moléculas de BT y TT.
Metodología
Los cálculos de estructura electrónica se efectuaron en una su percomputadora Origin 2000 Silicon Graphics con 18 proce sadores R10000, de la Universidad Autónoma Metropolitana, Campus Iztapalapa (México) con el paquete computacional Gaussian-98 [10]. Todas las geometrías se optimizaron sin restricciones de simetría al nivel de campo autoconsistente (SCF) Hartree-Fock Restringido (RHF) [11], con la base estandarizada 6-31G(d) [12]; dicho conjunto base incluye seis funciones Gaussianas primitivas de segundo orden para la descripción de átomos pesados (funciones d sobre los átomos de S y C). La convergencia utilizada en los cálculos fue el criterio de RMS density matrix = 1.00D-08.
Para el análisis conformacional de BT y TT, las geome trías se optimizaron a lo largo de la curva de potencial, abriendo los ángulos torsionales entre los planos de los anillos (θ y ϕ) en intervalos de 30° (Fig. 1), desde 0° (conformación cis planar) hasta 180° (conformación trans planar). Para determinar la localización de los mínimos gauche, se optimizaron otras dos geometrías en la vecindad de los confórmeros s-cis y s-trans. Las energías relativas se obtuvieron con respecto a la conformación de mínima energía.
Los valores para los potenciales de ionización vertical se calcularon de acuerdo con el teorema de Koopmans [13]. La energía de excitación corresponde a la diferencia de energías entre el orbital más alto ocupado (HOMO) y el orbital más bajo desocupado (LUMO). Las geometrías y densidad elec trónica se visualizaron con los paquetes computacionales Moldraw [14] y Molden [15], respectivamente.
Resultados y discusión
a. Análisis conformacional de 2,2'-bitiofeno (BT)
Geometrías
En la Tabla 1 se presentan los datos de las geometrías molecu lares optimizadas de los confórmeros más significativos encontrados para BT: s-cis planar, s-cis-gauche, perpendicular, s-trans-gauche, y s-trans planar. Para efectos de comparación, se incluyen las parámetros geométricos obtenidos por difracción electrónica en fase gas para los mínimos gauche [16]. En el caso de los parámetros interanulares C2 - C2' y C2' - C2 - C3, los valores estimados por el cálculo teórico son mayores que los valores experimentales en 0.7% para el mínimo s-cis-gauche y 2% para el mínimo s-trans-gauche. Las longitudes de enlace C2 C3, C3 - C4 y C4 - C5 son menores por 1.4%, mientras que las distancias S1 - C2 y S1 - C5 son mayores en 0.4%.
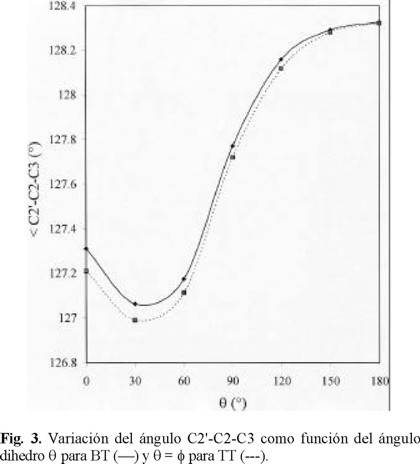
Debido a la rotación interna, los cambios más importantes se dan en los parámetros interanulares C2 - C2' y C2' - C2 -C3 (Tabla 1, Figs. 2 y 3). Estos cambios se originan en la evolución de los efectos de conjugación que ocurre entre las nubes electrónicas de los anillos, cuando pasan de una conformación planar a una perpendicular y en las interacciones de no enlace que están presentes en los confórmeros planares. En el caso del enlace C2 - C2', su longitud aumenta al pasar de las estructuras s-cis y s-trans planar a la conformación perpendicular, lo que podría estar relacionado directamente con la pérdida de conjugación ϖ que se da en esta última.

En el confórmero s-trans planar, el enlace C2 - C2' es más corto que el correspondiente en el confórmero s-cis planar, una posible consecuencia de la importancia que tienen las interacciones de no enlace en la forma s-cis. Para este rotámero, se encuentran presentes la interacción de no enlace entre los dobles enlaces C2 - C3 y C2'-C3', un contacto corto, H3 -H3', y la interacción de no enlace entre los pares libres de los átomos de azufre. La distancia S1 - S1' tiene un valor de 3.326 Å, el cual es menor a la suma de radios de van der Waals (3.60 Å) [17]. En el caso de la distancia H3 - H3', su valor es 2.403 Å, muy semejante al estimado de acuerdo a los radios de van der Waals (2.40 Å)[17].
En lo que respecta al confórmero s-trans planar, la distancia S1 - H3' tiene un valor de 2.928 Å, muy cercano al valor de la suma de radios de van der Waals (3.00 Å) [17]. Estas interacciones de no enlace permiten justificar que los enlaces interanulares S1 - C2 y C2 - C3 sean más largos para la conformación planar que para la estructura perpendicular.
Los ángulos de enlace S1 - C2 - C3 y C2 - C3 - H3 tienen, respectivamente, el menor y mayor valor para los confór meros planares, lo que permite atenuar el efecto de las interacciones de no enlace.
La longitud del enlace interanular C2 - C2' de la molécu la de BT es más corta que la correspondiente en la molécula de bifenilo [18], lo que indica una fuerte deslocalización elec trónica entre los dos anillos conjugados y sugiere un comportamiento poliénico. El valor obtenido para el enlace C2 - C2' en la conformación perpendicular es más pequeño que el cor respondiente enlace C - C en la molécula de etano [1], lo que puede atribuirse a efectos hiperconjugativos entre los electrones σ de un anillo y los electrones ϖ del otro [19].
Angulo torsional θ
Los cálculos predicen la existencia de un mínimo tipo gauche en la vecindad de los confórmeros s-cis planar y s-trans planar, en buen acuerdo con los datos experimentales provenientes de difracción electrónica [16]. El mínimo s-trans-gauche corresponde a un ángulo torsional de 147.79°. La estimación experimental por difracción electrónica predice un valor de 148 ± 3°. El valor experimental en la conformación s-cis-gauche es 35 ± 5°, mientras que el cálculo teórico predice que el ángulo torsional se encuentra en 42.33°, el cual es comparable con el máximo valor experimental.
Curva de energía potencial
La Tabla 2 contiene los valores de las energías relativas obtenidas para la rotación interna del BT como una función del ángulo dihedro θ. Con estos valores se obtuvo la curva de energía potencial mostrada en la figura 4. Las inflexiones presentes en la curva de potencial se deben a la presencia de los átomos de azufre que confieren gran importancia a las interacciones de no enlace en la región interanular y desestabilizan los confórmeros planares. Se observa que las conformaciones más estables tienen barreras menores a 1.6 kcal/mol y no son estructuras completamente planares.


La conformación más estable corresponde a un mínimo s-trans-gauche localizado a un ángulo torsional de 147.79° (ver Tabla 2). Un segundo mínimo local, s-cis-gauche, se presenta para un ángulo torsional de 42.33°. La máxima diferencia de energía entre dos conformaciones es de 1.930 kcal/mol, lo que sugiere una gran flexibilidad para la molécula de 2,2'-bitiofeno y justifica la variedad de conformaciones observadas experimentalmente para oligómeros de tiofeno.
El mínimo s-trans-gauche es 0.372 kcal/mol más estable que el confórmero s-trans planar, lo que se puede atribuir a que la interacción estérica entre los átomos de azufre e hidrógeno desestabiliza la conformación planar. Los confórmeros gauche están separados por una barrera de 0.68 kcal/mol. Este valor sugiere una fácil interconversión entre los dos mínimos gauche y permite explicar la coexistencia de los confórmeros s-cis y s-trans en fase gas [16] y en solución [20].
El mínimo s-cis-gauche es 1.25 kcal/mol más estable que el confórmero s-cis planar. Se espera que el confórmero s-cis planar sea menos estable debido a las interacciones de no en lace interanulares entre los pares libres de los átomos de azu fre y entre los dobles enlaces C2 - C3 y C2' - C3'. La curva de energía potencial establece que el confórmero s-cis planar corresponde al máximo absoluto del potencial, siendo menos estable 0.238 kcal/mol con respecto a la conformación perpendicular. El confórmero s-cis planar es menos estable por 1.558 kcal/mol con respecto al confórmero s-trans planar.
b. Análisis conformacional de 2,2':5',2"-tertiofeno (TT)
Geometrías
Los confórmeros más importantes encontrados para TT al variar los ángulos torsionales θ y ϕ corresponden a las estructuras cis-cis planar, cis-cis-gauche, perpendicular, trans-trans-gauche, cis-trans-planar, y trans-trans planar. Sus geometrías son no coplanares y presentan una distorsión fuera del plano que oscila entre 30 y 45°. En la Tabla 3 se presentan los datos de las geometrías moleculares optimizadas para la conformación trans-trans-gauche (149.4°, 149.4°).

Igual que en BT, la rotación interna da lugar a cambios significativos en los parámetros interanulares (Figs. 2 y 3). En el caso del enlace C2 - C2', su longitud aumenta cuando va de las estructuras s-cis y s-trans planar a la conformación perpendicular, debido a la pérdida de conjugación ϖ. El enlace C2 C2' es más corto en el confórmero cis-trans planar que en el cis- cis planar.
Angulos torsionales θ y ϕ
Los cálculos predicen que para las conformaciones trans-trans, el mínimo muestra dos ángulos dihedros con un valor absoluto de 149.4° (trans-trans-gauche); (41.6°,149°) para la conformación cis-trans-gauche y de (41.6°, 41.6°) en el caso del rotámero cis-cis-gauche. Estos valores están en buen acuerdo con los valores del ángulo torsional encontrado para BT, donde para las estructuras con giro trans y cis se calcularon los dos mínimos a 147.79° y 42.33°, respectivamente. Para evaluar la flexibilidad conformacional de TT y la inter conversión entre dos confórmeros, se estimaron las barreras de energía. Los resultados de estos cálculos se muestran en la Tabla 4. Las barreras obtenidas indican que también se puede esperar una gran flexibilidad conformacional para la molécula de TT.

La geometría trans-trans planar (θ = ϕ = 180°) corresponde a un máximo relativo de energía, que se encuentra solamente 0.668 kcal/mol por arriba del mínimo absoluto. Además, es conveniente hacer notar que las diferencias de energía calculadas posiblemente disminuyan cuando en los cálculos se incluyan efectos de correlación, tal y como sucede en el caso del BT [1].
Curva de energía potencial
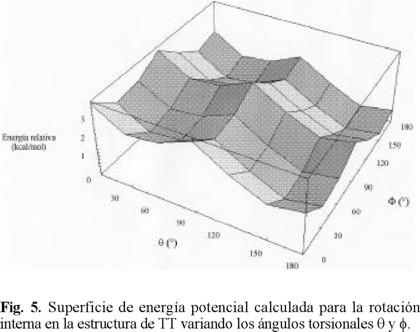
La figura 5 muestra la superficie de energía potencial calculada para TT al variar los ángulos torsionales θ y ϕ. Se observa que la conformación de mínima energía corresponde a la geometría molecular donde los anillos adyacentes están en una configuración trans con ángulos torsionales θ = ϕ = 149.4°. Tal y como sucede en la molécula de BT, la conformación cis-planar y la perpendicular son las menos estables (Tabla 4). La barrera de energía torsional entre las conformaciones totalmente planar y perpendicular es más alta que la observada para bitiofeno, debido a que la conjugación electrónica es mayor en el TT.
En la figura 6 se muestra la visualización de la densidad de carga en la molécula de TT. Se observa una mayor acumulación en los enlaces C = C y C - C del anillo central y en los enlaces C - C que conectan las unidades de tiofeno. La longitud de dichos enlaces puede ser un indicio de la conjugación existente en la estructura.

Al girar 90° los anillos de tiofeno (θ = 90°, ϕ = 90°), la conformación es desestabilizada por 2.93 kcal/mol. Las conformaciones s-cis de TT son más estables que las conformaciones perpendiculares, debido a la importancia de la conjugación electrónica.
c. Oligómeros con n = 4 - 8
La figura 7 muestra las geometrías optimizadas para los oligómeros n = 4 - 8. Se observa que a partir del oligómero de 6 unidades el ángulo torsional de la cadena tiene un valor de 31° entre los anillos adyacentes ubicados en los extremos y 29° entre los anillos internos. Esta flexibilidad encontrada para ca denas largas es acorde al comportamiento conformacional calculado para las moléculas de BT y TT.
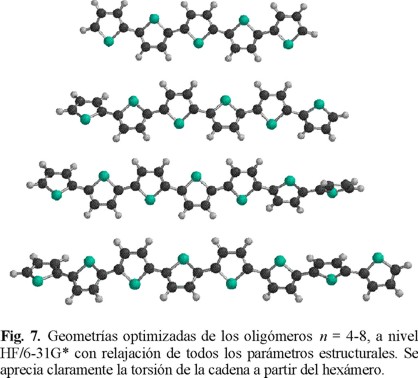
En lo que respecta a los enlaces C - C y C = C, se aprecia la alternancia a lo largo de la cadena, sugiriendo una estructu ra de tipo bencenoide que permite la deslocalización intramolecular de los electrones ϖ y apoya el modelo teórico que ba sado en este hecho intenta explicar las atractivas propiedades basadas en transferencia de carga que se observan en estos materiales. Esta alternancia se esquematiza en la figura 8.
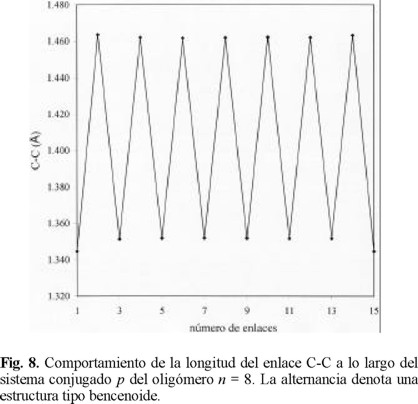
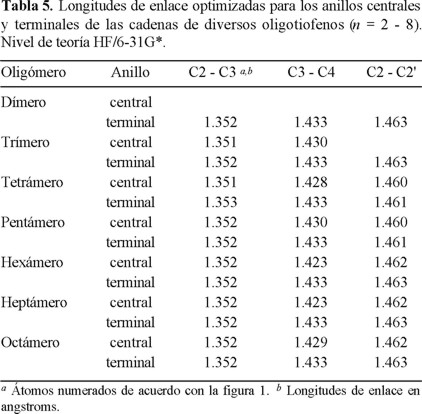
La Tabla 5 contiene los valores de los enlaces C - C considerando si el anillo es terminal o central en la cadena. El enlace C3 - C4, que es la base del anillo, es más corto cuando corresponde al anillo central. El enlace C2 - C3 mantiene un valor prácticamente constante de 1.35 Å, muy próximo al correspondiente a un enlace doble carbono - carbono. El enlace entre carbonos de las posiciones α de los anillos mantiene un valor constante de 1.46 Å a partir del oligómero con 6 unidades independientemente de corresponder a un anillo central o terminal de la cadena.
El Teorema de Koopmans [13] confiere significado físico al concepto de orbital molecular, asumiendo que la energía requerida para remover un electrón de una molécula es su potencial de ionización vertical (PIV) y lo define como el negativo del eigenvalor correspondiente al orbital más alto ocupado (HOMO). De acuerdo con este teorema, se obtuvieron los respectivos valores para los oligómeros.
Conforme aumenta la cadena, el PIV disminuye hasta llegar al pentámero, aumenta en el hexámero y a partir de ahí se mantiene prácticamente constante. Este cambio en la tendencia coincide con la aparición de torsión en la cadena.
Al incrementarse el número de anillos en la cadena, el HOMO es menos profundo (en términos de niveles energéticos) y la deslocalización a lo largo de la cadena aumenta. La torsión de la cadena disminuye la extensión de la conjugación y el HOMO es más profundo. La energía de excitación corresponde a la diferencia entre los eigenvalores del HOMO y LUMO; de acuerdo con la teoría de bandas del estado sólido, este parámetro constituye un indicio para medir y explicar la conductividad en estos materiales [21].
La tendencia de comportamiento de la energía de excitación es similar a la que presenta el PIV. Si se considera a la energía de excitación como un gap, al crecer la cadena existe mayor deslocalización y mayor conductividad. Los valores se reportan en la Tabla 6. Cabe mencionar que existe una sobre estimación de valores con respecto a los reportados experi mentalmente, pero la tendencia cualitativa apreciada es representativa.

Conclusiones
En la descripción de la estructura molecular de oligotiofenos se encuentran involucrados factores como el impedimento estérico entre átomos de azufre e hidrógeno de anillos adya centes, interacciones de no enlace y la conjugación electrónica ϖ entre los anillos.
Las curvas de energía potencial obtenidas para BT y TT justifican la gran variedad de conformaciones observadas experimentalmente para dichos oligómeros. En el caso de BT, la diferencia de energía entre los mínimos locales (confórmeros s-trans-gauche y s-cis-gauche) permite explicar la evidencia experimental de su semejanza en abundancia relativa.
Los oligómeros muestran una estructura bencenoide. A partir de 6 anillos se observa torsión en la cadena y un valor prácticamente constante para el PIV y la energía de excitación. La tendencia observada permite proponer al hexámero como modelo de comportamiento representativo, cuyo estudio detallado cualitativo y cuantitativo con diversas metodologías, permitirá comprender las peculiaridades estructurales y electrónicas que controlan el transporte de carga en politiofenos.
Agradecimientos
A. R. agradece al Consejo Nacional de Ciencia y Tecnología la beca otorgada para la realización de estudios de Doctorado en Ciencias.
Referencias
1. Ortí, E.; Viruela, P.; Sánchez-Marín, J.; Tomás, F. J. Phys. Chem. 1995, 99, 4955-4963. [ Links ]
2. Inganäs, O.; Salaneck, W.R.; Österholm, J.-E.; Laakso, J. Synth. Met. 1988, 22, 395-406. [ Links ]
3. Salaneck, W.R.; Inganäs, O.; Thémans, B.; Nilsson, J.O.; Sjögren, B.; Österholm, J.-E.; Brédas, J.L.; Svensson, S. J. Chem. Phys. 1988, 89, 4613-4619. [ Links ]
4. Foresman, J.; Frisch A., Exploring Chemistry with Electronic Structure Methods, Second Edition, 1995-96, Gaussian, Inc. [ Links ]
5. Fujimoto H., Nagashima, U., Inokuchi H., Seki, K., Cao, Y. Nakahara, H., Nakayama, J., Hoshino, M., Fukuda, K. J. Chem. Phys. 1990, 92, 4077-4091. [ Links ]
6. Degli, A., Zerbetto, F. J. Phys. Chem A 1997, 101, 7283-7291. [ Links ]
7. Alemán, C., Julià, L. J. Phys. Chem. 1996, 100, 14661-14664. [ Links ]
8. Viruela, P., Viruela, R., Orti, E. Int. Journal of Quantum Chemis-try 1998, 70, 303-312. [ Links ]
9. Brèdas, J. L., "Electronic Structure of Highly Conducting Polymers" Chapter 25 in Handbook of Conducting Polymers vol. 2, Ed. Terje A. Skotheim, Marcel Dekker, Inc., 1986. [ Links ]
10. Gaussian 98, Revision A.6,M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, and J. A. Pople. [ Links ]
11. Roothan, C.C.J. Rev. Mod. Phys. 1951, 23, 69-89. [ Links ]
12. Ditchfield, R.; Hehre, W.J.; Pople, A.J. J. Chem. Phys. 1971, 54, 724-728. [ Links ]
13. Koopmans, T.A. Physica 1933, 1, 104-113. [ Links ]
14. MOLDRAW 1.0 Ver. G; Programa de modelado molecular para PC, ambiente Windows 3.1, 95; P. Ugliengo, D. Viterbo y G. Chiari. [ Links ]
15. MOLDEN: Paquete computacional para visualización de densidades moleculares, plataforma UNIX; G. Schaftenaar. [ Links ]
16. Samdal, S.; Samuelsen, E.J.; Volden, H.V. Synth. Met. 1993, 59, 259-265. [ Links ]
17. Para la estimación de las sumas de radios de van der Waals se tomaron los valores reportados por Cruz, D., Chamizo, J. A., Garritz, A., "Estructura atómica, un enfoque químico", Addison-Wesley Iberoamericana, 1991, págs. 738-739. [ Links ]
18. Charbonneau, G.; Delugeard, V. Acta Crystallogr. B 1976, 32, 1420-1423. [ Links ]
19. Daudey, J.P.; Trinquier, G.; Barthelat, .C.; Malrieu, J.P. Tetrahedron 1980, 36, 3399-3401. [ Links ]
20. Berardi, R.; Spinozzi, F.; Zannoni, C. Liq. Cryst. 1994, 16, 381. [ Links ]
21. Bakhshi, A.K. Materials Science and Engineering C 1995, 3, 249-255. [ Links ]

