










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.44 n.1 Ciudad de México Jan./Mar. 2000
Investigación
A Study of Cooperatively Dimerizing Hard Spheres in a Cylindrical Pore by Using Grand Canonical Monte Carlo Simulation
Lorena Romero-Salazar,1* Wojciech Rzysko,2,3 Orest Pizio,1,a Stefan Sokolowsky 3
1 Facultad de Ciencias de la UAEMéx., Av. Inst. Literario 100, Toluca 50000, México.
2 Instituto de Química de la UNAM, Ciudad Universitaria, Circuito Exterior, Coyoacán 04510, México D.F.
3 Department for the Modelling of Physico-Chemical Processes, Marie Curie-Sklodowska University, Lublin 200-31, Poland.
a On sabbatical leave from: Instituto de Química de la UNAM.
Recibido el 14 de enero del 2000.
Aceptado el 24 de febrero del 2000.
Abstract
We have investigated a hard sphere fluid adsorbed in a cylindrical pore with molecularly rough walls by using grand canonical Monte Carlo simulations. The Hamiltonian of the interparticle interactions includes a three body term promoting dimerization of adsorbed species in the vicinity of a given solid atom belonging to the pore walls, such that the chemical association mechanism is cooperative. We have obtained the adsorption isotherms and the density profiles of fluid particles in the pore. It is shown that the microporosity of molecularly rough walls influences the fraction of dimerized species in competition with the effect of association energy.
Key words: Gran Canonical Monte Carlo simulation; adsorption, chemical association, cylindrical pore.
Resumen
Utilizando simulaciones de Monte Carlo en el ensamble Gran Canónico, se estudia un fluido de esferas duras adsorbido en un poro cilíndrico con paredes rugosas. En la contribución de interacciones del Hamiltoniano del sistema se incluye un término de tres cuerpos, tal que dada una partícula sólida de la pared del poro, ésta favorece la dimerización de la especie que es adsorbida en su vecindad, es decir, el mecanismo de asociación es un efecto cooperativo. Se presentan las isotermas de adsorción y los correspondientes perfiles de densidad de las partículas del fluido en el poro. Se muestra cómo la microporosidad y la rugosidad de la pared influyen en la fracción de la especie dimerizada en competencia con los efectos de la energía de asociación.
Palabras clave: Simulación de Monte Carlo Gran Canónica, adsorción, asociación química, poro cilíndrico.
Dedicated to the memory of Prof. Raul Cetina Rosado
Introduction
The structural and thermodynamic properties, including phase behavior, of fluids confined in pores and in microporous media are of much interest for basic research and for several applications. This is the reason why the adsorption of simple fluids in pores with even a highly idealized geometry, such as the slit-like, the cylindrical and spherical shape, has been the subject of several studies. The methods permitting microscopic description of adsorption in the multiple pore systems dependent on the shape of a single, individual pore are well developed, see e.g. Ref. [1]. Tubular pores represent an important class of pores for practical applications. Specifically, one group of synthesized mesoporous silicates known as MCM-41 is thought to consist of hexagonal arrays of parallel tubular (almost cylindrical) pores separated by amorhous silica [2]. Several attempts to characterize with precision a microporous structure of these materials have been undertaken. In particular, Edler et al. [2] have concluded that both the model of cylindrical pores with "hairy" rough, microporous walls and the model of cylindrical pores interconnected by narrower micropore pathways are appropriate to describe the experimental adsorption isotherms of hydrogen in aforementioned silicates.
The development of adequate microscopic modelling for cylindrical pores with molecularly rough walls is not straight-forward. Moreover it is not clear in advance what kind of effects would yield surface roughness of the pore walls on thermodynamic properties of adsorbed fluids. Undoubtedly, it is most convenient to apply computer simulation methods, see e.g. [3], to study the problem rather than to attempt the development of approximate theoretical approaches. We model "hairy" microporous walls of a cylinder by randomly depositing fluid species (they will be called matrix species in what follows) in a layer adjacent ot the cylindrical pore walls. This type of modelling for simulation purposes has been exploited in some other aspect in recent works from our laboratory [4]. To summarize these comments, our interest in the present work is to study the adsorption of fluids in a cylindrical pore with molecularly rough, microporous walls. However, our interest is in a particular model for a confined fluid.
The research projects of our laboratory during last years have been focused in inhomogeneous associating fluids. In particular, the behavior of dimerizing, chain-forming and network-forming models under semiinfinite confinement and in slit-like pores have been studied, see e.g. [5, 6]. The formation of bonds between monomeric units has been generated by the introduction either of a square well attractive interaction shell into the hard core of particles (the model of Cummnigs and Stell [7, 8]) or of a square well attractive bonding sites providing orientational bonding (the models of Wertheim [9, 10]). However, the attractive, associative interactions have been assumed pairwise, as in common liquid state theory.Also the possibility of the formation of bonds between a fluid species and surface atoms alone, or in competition with bonding between solely fluid species, has been analyzed [11].
On the other hand, very recently it has been shown that the cooperativity of bonding (in other words, the dependence of the formation of a bond between a pair of fluid particles on the proximity of the third neighbor) results in a very unusual thermodynamic behavior of associating, homogeneous model fluids [12-14]. However, performed analysis of the effects of the cooperativity of bonding has been restricted to lattice models and to homogeneous situation [15], i.e. to the bulk fluids.
In this preliminary report, out objective is to introduce a microscopic model for a cooperative bonding at a very simple level, providing however, a path to a more sophisticated future computer simulation study. Our modelling has been influenced by the Langmuir-Hinshelwood mechanism for surface chemical reactions. Namely, this mechanism assumes that two particles can react between themselves with the formation of a product, if both of them have been adsorbed on a solid surface. We would like to reformulate this fundamental issue by assuming that two fluid particles adsorbed into a cylindrical pore can react with the formation of a bonded complex, if both of them are located sufficiently close to a matrix particle, that is located in a microporous hairy layer of the cylindrical pore walls. In other words, the microporous disordered matrix layer serves as a substrate for a cooperative chemical association. The model, however, will be discussed in more detail in the following section.
We would like to conclude the introductory section by few comments. First, as usual in studies of adsorption, one needs to perform simulation in the grand canonical ensamble ought to the fact that the equilibrium between the bulk fluid and the adsorbed species in the pore takes place at constant chemical potential. The simulation of chemically associating fluids even in canonical ensamble is not a simple task because the bonding configurations usually represent a small part of the volume of the phase space. Several "tricks" are necessary to provide adequate sampling of configurations, see e.g. [16]. Second, the model in question in the present study is a very simple one. However, it includes two important novel elements. namely it includes the effect of the pore walls roughness at a microscopic scale and the effect of cooperativity in chemical association. These two elements are indispensable for future development of the studies of adsorption that represents a preliminary stage of heterogeneous catalysis.
The model and procedure
We consider a cylindrical pore consisting of two coaxial cylinders with the radii, Rcl = 5 and Rcs = 3. The exterior of the larger cylinder is impermeable to either matrix or fluid species. It is assumed that matrix hard sphere particles with unit diameter, σm = 1, have been deposited in some way in the volume restricted by the radii of larger and smaller cylinder, i.e. in the space Rcs < Rm,i < Rcl, where R2m,i = x2m,i + y2m,i is the square of the radial vector characterizing the position of the i-th particle belonging to matrix species, with respect to the axis of the cylindrical pore in question.
The first stage of the computer experiment is performed as follows. The centers of matrix spheres have been distributed randomly in the space between inner and outer cylinders with the radii, Rcs and Rcl. However, overlapping configurations of matrix spherical cores have been excluded. Height of both cylinders is lz. In what follows the periodic boundary conditions along z axis are used. The matrix species do not interact with the walls of outer cylinder. The inner cylinder is just an imaginary boundary for the coordinates of matrix species. Our intention in the distribution of matrix species has not been to obtain any varying density profile for them. Rather we intended to obtain a uniform distribution of hard spheres between two cylinders with the average density for matrix species, ρm = Nm/[π (R2cl − R2cs)lz]. The volume between large and small cylinder can be characterized by the microporosity value, p. We define the microporosity of that space as follows, p = 1 − Nm/{6[(Rcl + 0.5)2 − (Rcs − 0.5)2]lz}. A configuration of matrix particles has been rigidly fixed before performing simulation of adsorption of fluid hard spheres into the pore in the grand canonical ensemble.
Now, we would like to perform a simulation experiment for the adsorption of a fluid into the pore with prepared micro-porous walls. For simplicity, at this preliminary stage of a larger project, we consider a hard sphere fluid of particles with the diameter σf. Without loss of much generality the diameter of fluid species is chosen equal to the one of matrix atoms, i.e. σf = σm = 1. The fluid-matrix interactions are assumed additive, σfm = (σf + σm)/2. The centers of fluid particles are restricted to the wider cylinder volume, i.e. the fluid-pore interaction is the following:
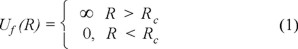
where R is the distance along the normal of a fluid particle with respect to the pore walls.
Essential element of our modelling is that, we assume that there is a three-body interaction, Uffm (r1,r2,rim), where the first two arguments describe coordinates of fluid particles, whereas the third, rim, describes the coordinate of i-th particle belonging to matrix species, m. This interaction is chosen in the form
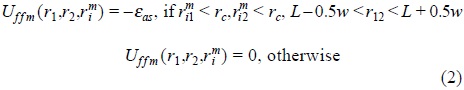
where, rmi1 = | rim − r1 | , L is the bonding length, w is the width of associative square well, εas is the association energy; rc is the range of a spherical shell around a matrix particle in which fluid species can associate. In addition, we have included also the fourth-order associative potential of the type, Uffm (r1,r2,rim,rjm),
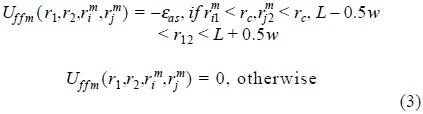
which promotes dimerization of fluid species, if each of the monomers, 1 and 2 are located sufficiently close to two matrix particles with the coordinates, rim and rjm.
The model for associative interaction is the extension of the primitive model for chemical association in bulk fluids developed and well studied by Cummings and Stell [7, 8]. Geometry of associative interaction in the form chosen in this study does not prohibit formally the formation of a higher-order associates, see e.g. Ref.[8] for detailed discussion. However, fraction of these species is very low and therefore will be neglected.
The fluid species in the bulk is characterized by the chemical potential βµ f . The fluid particles are adsorbed in the entire large cylinder and attain in equilibrium the density distribution characterized by the density profile ρf (R ). The density profile describes the structure of adsorbed fluid. Moreover, by using either ρf (R) or the number of adsorbed fluid particles in the simulation, < Nf >, we obtain the normalized adsorption isotherms, ρ f (βµ f , p),
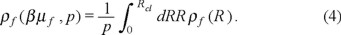
Normalization of the adsorption isotherms has been performed to facilitate a comparison of adsorption of a fluid in pores with different microporosity.
The Monte Carlo simulations in the grand canonical ensemble has been performed as common [17]. Each simulational step have consisted of attempts to displace a fluid particle, as in common Metropolis MC algorythm, to delete a randomly chosen particle or to create a particle at a random position inside an outer cylinder. The displacement scheme has been usual [17], a more sophisticated displacement scheme will be used in a future study of the models with directional associative forces. The acceptance of particles displacements has not been lower than 35%-40% in all runs. The productive part of each of the runs had ranged from 5 × 105 to 106 steps dependent on the chemical potential of fluid species, preceeded by equilibration during 105 steps.
Results and discussion
Let us proceed with the description of the results obtained. All the simulation have been performed at two values for association energy βεas = 1, and βεas = 5, and several values for the chemical potential in each case. We discuss first the adsorption isotherms and then proceed with the description of the density profiles of fluid particles as well as the density profiles of monomers and dimers.
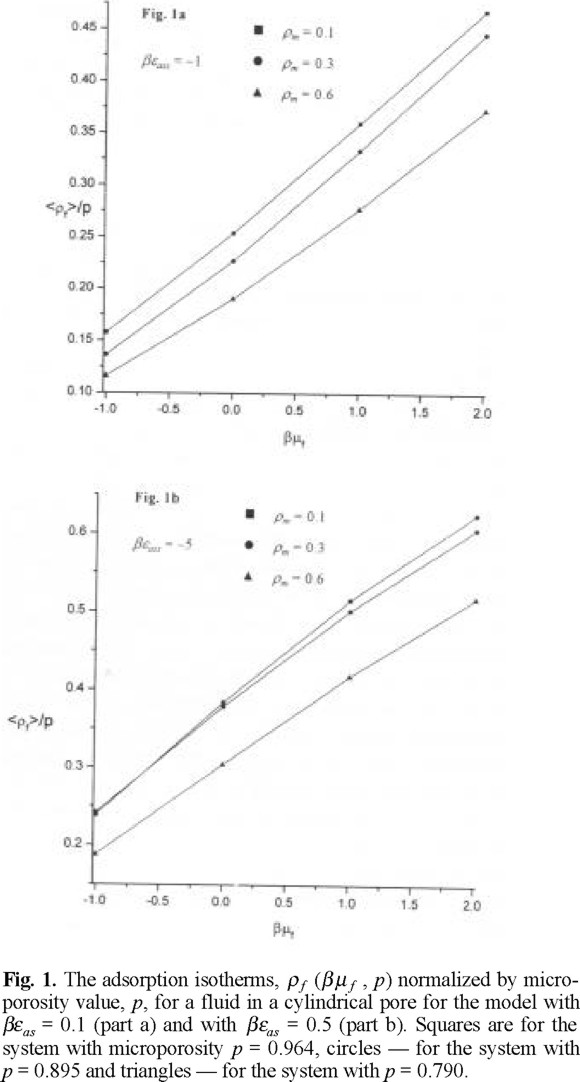
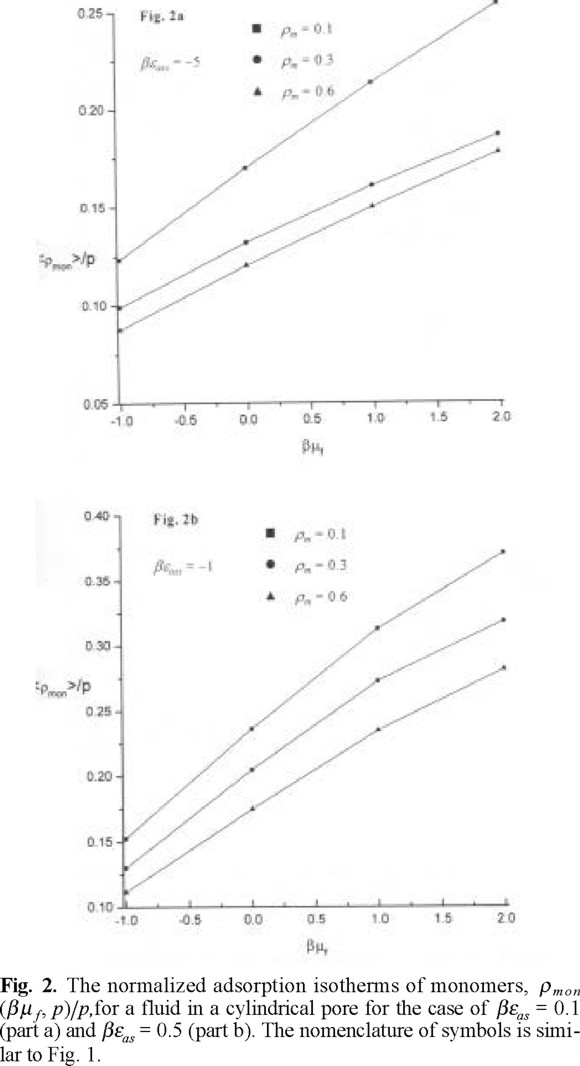
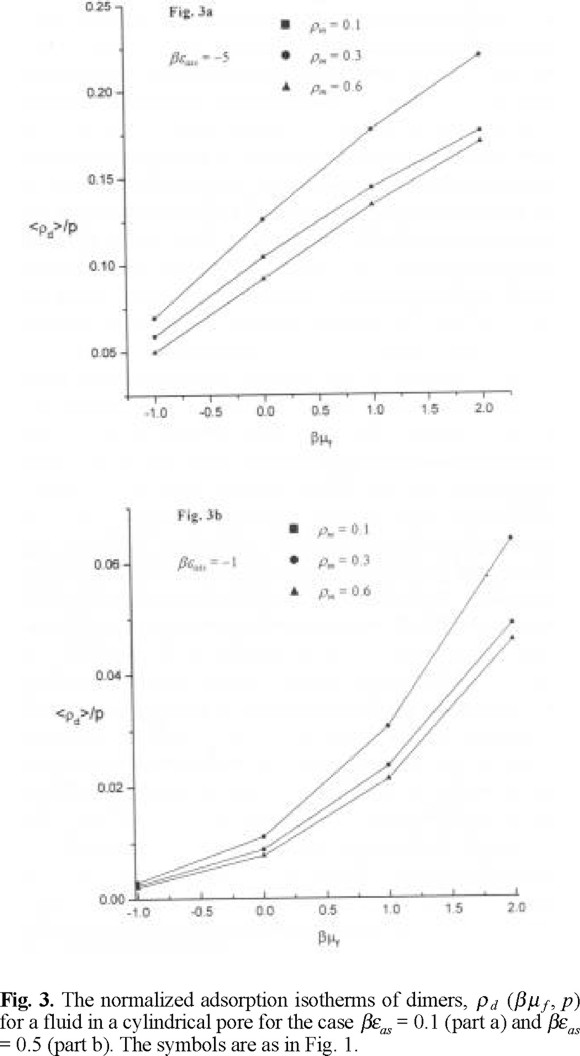
The adsorption isotherms describing adsorbed density normalized by microporosity versus chemical potential of a hard sphere fluid are shown in Figs. 1a and 1b, for βεas = 1 and for βεas = 5, respectively. The adsorbed density increases with increasing chemical potential, as expected. The adsorbed density is lower for the case when matrix film is characterized by a low microporosity or in other words if the matrix film is more dense, see Fig. 1a. The adsorbed density is higher if the association energy between species is higher. This behavior can be attributed to a stronger penetration of fluid species into a microporous matrix medium leaving more space for fluid species to adsorb into a central part of the pore. One, important, in our opinion, observation concerns with the adsorption at a higher association energy, βεas = 5. We observe a crossover in the behavior of the adsorption isotherms at matrix density ρm = 0.1 (p = 0.964) and ρm = 0.3 (p = 0.895), at a chemical potential β µf ≈ −0.75. To get a better insight into the nature of this crossover, we have calculated the adorption isotherms for monomers and dimers separately. The adsorption isotherms for monomers do not exhibit unusual features, Figs. 2a and 2b. The density of monomers monotonously increases with increasing chemical potential. The density of monomers at a given chemical potential is lower for the model with higher association energy, βεas = 5. As expected, the adsorbed density of monomers is lower in the case of a low microporosity of matrix film. However, the adsorbed density of dimerized species exhibits interesting trends, Figs. 3a and 3b. Both, for a lower and higher value of association energy, βεas = 1, and βεas = 5, the adsorbed density of dimers is the lowest for a low microporosity of matrix film, i.e. for the highest density of matrix species, ρm = 0.6 (p = 0.790). Evidently, the dimer species in this case may be predominantly formed only on the external surface of the matrix film in the region of distances Rcs. Due to low microporosity in this case, the fluid species do not penetrate into a matrix film. For a higher microporosity of the matrix film, i.e. for a lower density, ρm = 0.3 we observe that the density of dimers is higher along all the range of the chemical potentials studied. For even higher microporosity of matrix layer, i.e. at ρm = 0.1 the dimer density decreases because of a lower number of matrix particles promoting association, compared to the case ρm = 0.3. However, at a given chemical potential, the dimer density remains higher than for the case ρm = 0.6. This nonlinearity in the dependence of the dimer density on microporosity of matrix layer promoting chemical association is one of our main findings. The density profiles of adsorbed species provide another interesting view in the emerging picture.
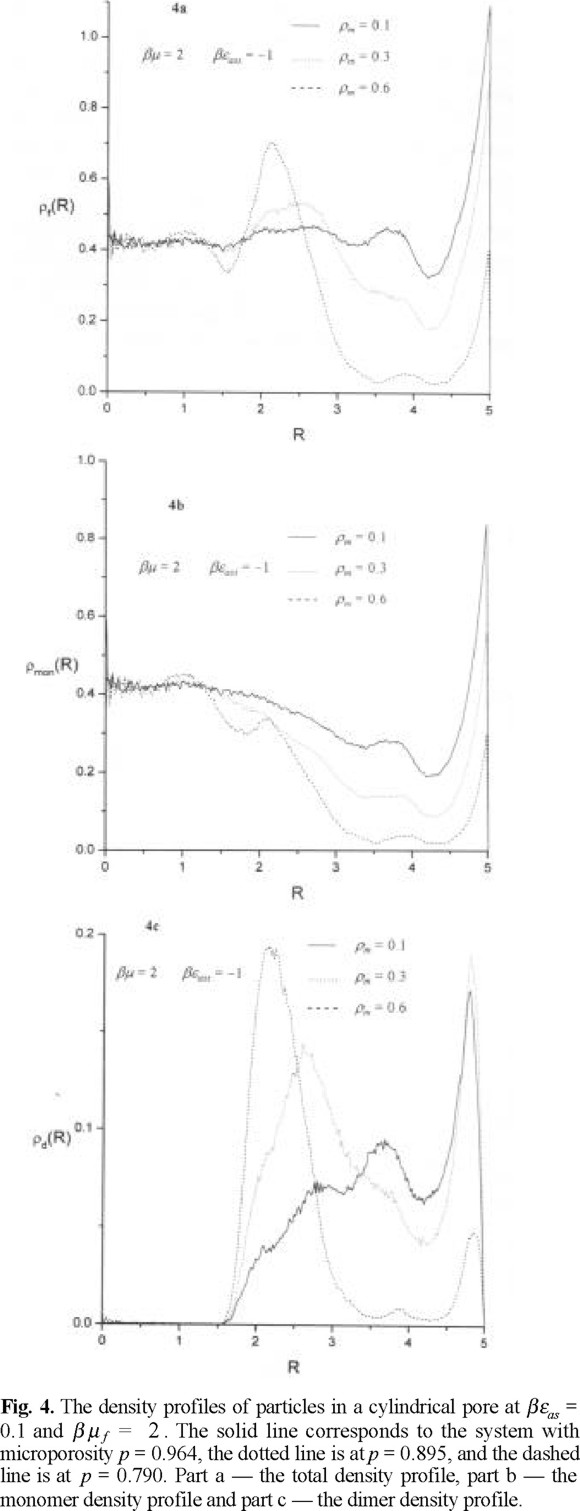
The evolution of the density profiles of fluid particles with microporosity, at βµ f = 2 and βεas = 1 is shown in Figs. 4a-4c. We observe that the density profile of fluid particles, ρ (r), in the case of a highly microporous matrix layer behaves quite similar to a hard sphere profile. There is a maximum of hard sphere density in contact with a wall of an outer cylinder and the density profile exhibits oscillatory behavior, Fig.4a. The contact value decreases with decreasing microporosity such that a maximum of fluid density develops close to the external surface of matrix layer. However, this density profile is not entirely determined by the profile of monomer density (Fig.4b). The monomer density profile exhibits similarity to the total density profile, ρ (r), only in the vicinity of the walls of the outer cylinder. Next, in the body of the matrix layer the density of monomers is quite low with trends to increase outside of the matrix layer. In contrast, the density profile of the centers of dimers, ρd (r), manifest strong trends for layering. The dimers preferentially form close to the outer cylinder surface in the case of a highly microporous matrix layer. In the case of a lower microporosity of the matrix layer we observe a fraction of them formed close to the surface of the outer cylinder but most of them begin to form in the region close to the exterior of the matrix layer, i.e. closer to the pore interior. Similar trends we observe at βµ f = 2 and βεas = 5, see Figs. 5a-5c. The main conclusion following from these density profiles is that the dimer distribution mostly determines the overall density behavior in the matrix layer body and close to the external surface of matrix layer. On the other hand, the density profile of monomers contribute to the overall density close to the outer cylinder surface and in the central part of the pore.
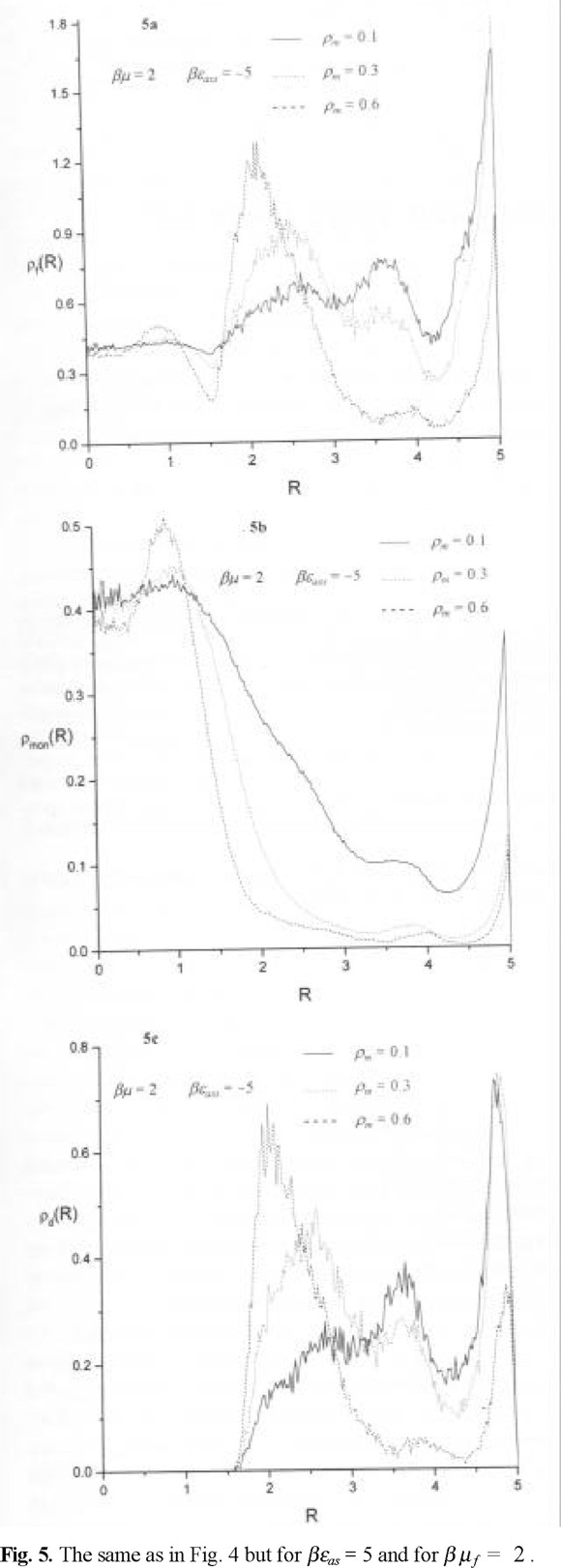
To conclude, we have considered adsorption of a hard sphere fluid into a cylindrical pore with the walls covered by disordered matrix layer. Our main focus is in determining the density of dimer species that can be formed in this layer via chemical association mechanism. Association is generated by a many-body potential involving two fluid particles and matrix species. The model has been studied by using GCMC simulation. The development of theoretical approaches for the model in question seems difficult at present, and anyway would require simulation data to test. The model in question mimickes, in a simplified manner, thermodynamic aspects of heterogeneous catalytic action of matrix species in a cylindrical pore. However, several interesting extensions easily can be seen, if the model for interparticle nonassociative and associative interactions, as well as for the interaction between species and pore walls would be more sophisticated.
Acknowledgements
This project has been supported by the National Council for Science and Technology (CONACyT), grants No.25301-E and No.I25363-E. L.R-S. acknowledges the access to computer facilities at ININ-Mexico. We are grateful to the Silicon Graphics Inc. for support of this project as well.
References
1. Thomas, J.M.; Thomas, W.J. Principles and Practice of Heterogeneous Catalysis, VCH, Weinheim, 1997, 257-313. [ Links ]
2. Edler K.J.; Reynolds, P.A.; Brandon P.J.; Trouw, F.R.; White J.W. J. Chem. Soc. Faraday Trans. 1997, 93, 1667-1676. [ Links ]
3. Schoen M. Computer Simulations of Condensed Phases in Complex Geometry, Ed. Springer Verlag, Berlin-Heidenberg, 1993, 75-92. [ Links ]
4. Kovalenko, A.; Sokolowski, S.; Henderson, D.; Pizio, O. Phys. Rev. E 1998, 57, 1824-1834. [ Links ]
5. Huerta, A; Sokolowski, S.; Pizio, O. Mol. Phys. 1999, 97, 919-930. [ Links ]
6. Huerta, A; Sokolowski, S.; Pizio, O. J. Chem. Phys. 2000, 12, 4286-4293. [ Links ]
7. Cummings, P.T.; Stell, G. Mol. Phys. 1984, 51, 253-287. [ Links ]
8. Cummings, P.T.; Stell, G. Mol. Phys. 1985, 55, 33-48. [ Links ]
9. Wertheim, M.S., J. Stat. Phys. 1984, 35, 19-34; 35-49. [ Links ]
10. Wertheim, M.S., J. Stat. Phys. 1984, 42, 459-476; 477-491. [ Links ]
11. Pizio, O.; Sokolowski, S. Phys. Rev. E 1997, 56, R63-R66. [ Links ]
12. Roberts C.J.; Debenedetti, P.G. J. Chem. Phys. 1996, 105, 658-672. [ Links ]
13. Roberts C.J.; Panagiotopulos A. Z.; Debenedetti, P.G. Phys. Rev. Lett. 1996, 77, 4386-4389. [ Links ]
14. Patrykiejew, A.; Sokolowski, S.; Pizio, O. Phys. Rev. Lett. 1999, 83, 3442-3445. [ Links ]
15. Sear, R.; Jackson, G. J. Chem. Phys. 1996, 105, 1113-1120. [ Links ]
16. Duda, Yu.; Pizio, O.; Sokolowski, S.; Bryk, P. J. Phys. Chem. B, 1998, 102, 5094-5494. [ Links ]
17. Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, Ed. Claredon Press, Oxford, 1986, 110-135. [ Links ]

