










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista latinoamericana de química
versão impressa ISSN 0370-5943
Rev. latinoam. quím vol.41 no.2 Naucalpan de Juárez Ago. 2013
Advances in computational approaches for drug discovery based on natural products
José L. Medina-Franco*
Torrey Pines Institute for Molecular Studies, 11350 SW Village Parkway, Port St. Lucie, Florida 34987, USA
*Corresponding author:
Phone: +1-772-345-4685, Fax: +1-772-345-3649,,
E-mail: jose.medina.franco@gmail.com.
Current address: Mayo Clinic, Scottsdale,
Arizona 85259, USA.
-mail: MedinaFranco.Jose@mayo.edu
Received May 2013.
Accepted July 2013.
ABSTRACT
Drug discovery based on natural products has a long successful history. To further advance the identification of new drugs from compounds of natural origin, natural product research is increasingly being combined with computer-aided drug design techniques. Herein, we review the recent advances in the application of chemoinformatics methods to quantify the chemical diversity and structural complexity of natural products and analyze their distribution in chemical space. We also discuss the progress in virtual screening to systematically identify bioactive compounds in natural products databases and the advancement of target fishing methods to uncover molecular targets of compounds from natural origin.
Key words: chemical space, chemoinformatics, compound databases, diversity analysis, docking, molecular modeling, target fishing, virtual screening.
RESUMEN
La identificación de fármacos basado en productos naturales tiene una larga historia de éxitos. Para incrementar el progreso del descubrimiento de fármacos de compuestos de origen natural, la investigación de productos naturales se está integrando cada vez más con técnicas empleadas en diseño de fármacos asistido por computadora. En este trabajo se hace una revisión de los avances recientes en la aplicación de métodos quimionformáticos para cuantificar la diversidad química y complejidad estructural de productos naturales y su distribución en el espacio químico. También se discute el progreso en el cribado virtual para identificar en forma sistemática compuestos bioactivos en bases de datos de productos naturales y métodos de búsqueda de dianas moleculares para revelar blancos moleculares de compuestos de origen natural.
Palabras clave: espacio químico, quimioinformática, bases de datos moleculares, análisis de la diversidad, acomplamiento molecular, modelado molecular, cribado virtual de dianas moleculares, cribado virtual de compuestos.
INTRODUCTION
Computational approaches commonly used in computer-aided drug design (CADD) have made significant contributions to the different stages of drug discovery. Advances in this filed have been reviewed recently in a number of publications (Xiang et al., 2012, Chen et al., 2012c, Ou-Yang et al., 2012). CADD includes several methodologies that can be classified in two major groups depending on the availability of the three dimensional coordinates of the target (Medina-Franco et al., 2006), namely structure-based and ligand-based approaches. The reader is referred to recent reviews of the contributions of specific computational approaches to drug discovery including molecular dynamics (Durrant and Mc-Cammon, 2011), pharmacophore modeling (Sanders et al., 2012), chemoinformatics (Duffy et al., 2012), treatment of receptor flexibility to model biomolecular recognition (Sinko et al. , 2013) and small molecule and protein-protein docking (Yuriev and Ramsland, 2013, Bienstock, 2012). Successful contributions of CADD to research projects have been encouraged by the increasing number of software, databases, and online tools available for medicinal chemists, biologists, and the research community in general (Liao et al., 2011).
Natural products have a rich history in drug identification and development (Newman, 2008, Ganesan, 2008, Lachance et al., 2012). For a long time, around 80% of drugs found their sources directly in natural products or compounds inspired by natural sources. It has been reported that, since 1994, 50% of the approved drugs have roots on natural products (Clark et al., 2010, Li and Vederas, 2009). The broader coverage of chemical space of natural compounds as compared to synthetic molecules gives an advantage to the former to identify novel structural classes (Harvey, 2008, Bohlin et al., 2010). Traditionally, identifying active compounds from natural products rely on the experimental evaluation of natural products in a set of biological assays available. Despite the fact that this approach has given rise to the successful identification of lead compounds and approved drugs discussed above, it is anticipated that combining computational approaches with experimental-based natural product research will enhance the success rate. In this regard, Barlow et al. reviewed the integration of in silico studies with Chinese herbal medicine's research (Barlow et al. , 2012). The synergy between other well-established drug discovery approaches such as virtual screening and combinatorial chemistry have been discussed elsewhere (López-Vallejo et al., 2011).
In this manuscript, we review the advances in the integration of CADD with natural products research. The review is organized in two major sections. The first one is focused on computational analysis of natural products databases using chemoinformatics methods. This section covers sources of compound databases and summarizes recent examples of the quantitative measure of structural diversity, complexity, profile of physicochemical properties and distribution in chemical space of natural products. The second section is dedicated to the progress of computational approaches for natural product-based drug discovery with emphasis on virtual screening to identify active molecules for molecular targets and target fishing to uncover putative targets for natural products.
CHEMOINFORMATIC APPROACHES
'Chemoinformatics' also called 'cheminformatics' or 'chemical information science' has various definitions, for example, 'the application of informatic methods to solve chemical problems' (Engel, 2006). Varnek and Baskin (Varnek and Baskin, 2011) and Willet (Willett, 2011) reviewed other definitions. Chemoinformatics comprises a plethora of computational techniques to organize, mine, visualize, and analyze the diversity and coverage of the chemical space of compound collections. The reader is referred to other reviews of chemoinformatic methods commonly used in the industry, academia and other research groups for lead identification and development and analysis of chemical databases (Duffy et al., 2012, Medina-Franco, 2013). In the next following sub-sections we will review the progress of major chemoinformatic-related areas of natural product research focused on lead identification.
Chemical databases of natural products
Compound databases represent a major source for storage, mining and sharing of chemical information, in some cases, including biological information. The source and impact on chemical databases on drug discovery is extensively discussed elsewhere (Scior et al., 2007, Barbosa and Del Rio, 2012). Clark et al. published diverse initiatives developed in various countries including France, Australia and Japan to foster drug discovery collaborations with academic groups (Clark et al., 2010). In addition to commercial sources of compounds for computational screening there are publicly available large compounds databases annotated with biological activity. Perhaps the most representative examples are PubChem, ChEMBL and Binding Database (Nicola et al., 2012).
Yongye and Medina-Franco recently compiled a list of five natural products databases whose structures are readily accessible on the web (Yongye et al., 2012). Such databases contain between 560 and 89000 compounds and the numbers are growing. The large and commonly used ZINC database (at the time of writing, May 2013, it contains over 19 million molecules) includes major subsets of natural products (Irwin and Shoichet, 2005).
The Traditional Chinese Medicine (TCM) database is one of the major sources of natural products freely available online (Chen, 2011). This database has been extensively analyzed in terms of physicochemical properties and chemical space coverage (see below) (López-Vallejo et al., 2012). Based on this database, the cloud-computing system iScreen was developed. This is a web server for docking TCM followed by customized de novo drug design (Tsai et al., 2011). iScreen is available at http://iScreen.cmu.edu.tw/. TCM has been used successfully to identify pancreatic triacylglycerol lipase inhibitors using in-silico approaches (Chen et al., 2012b).
A second major source of natural products freely available online is the Universal Natural Products Database (UNPD) developed by Gu et. al (Gu et al., 2013). UNPD available at http://pkuxxj.pku.edu.cn/ UNPD is comprised of 197201 compounds obtained from plants, animals and microorganisms. The physicochemical properties of this database have been analyzed. The physicochemical properties were employed as a basis to generate a visual comparison of the chemical space covered by UNPD and drugs concluding that there is a large overlap (Gu et al., 2013).
In Mexico, Esquivel and colleagues at the Informatics Unit of the Chemistry Institute of the National Autonomous University of Mexico (UNIIQUIM / UNAM for the name in Spanish) is building a comprehensive database of natural products that have been published by the Chemistry Institute of UNAM. It is estimated that the database will have information for more than 3000 chemical substances isolated and characterized. The database is freely searchable at http://uniiquim.iquimica.unam.mx.
In Brazil, Valli et al. developed the NuBBE database which is a web-based database available at http://nubbe.iq.unesp.br/nubbeDB.html that includes secondary metabolites and derivatives from Brazil (Valli et al. , 2013). Currently, the database contains 640 compounds collected from 170 scientific publications by the Nuclei of Bioassays, Biosynthesis and Ecophysiology of Natural Products (NuBBE) group. The database will be constantly updated with upcoming information (Valli et al., 2013).
Table 1 summarizes selected chemical databases of natural products that can be searched online or whose structures can be downloaded. The table focuses on collections recently published. Other compound collections are comprehensively reviewed elsewhere (Clark et al., 2010, Barlow et al., 2012, Yongye et al., 2012).
Diversity and structural complexity analysis
Quantitative analysis of the structural diversity of compound databases is important because it provides insights to prioritize library or sub-library selection for experimental screening. In particular, diversity analysis helps to assess the structural novelty of a compound collection (Medina-Franco, 2012). If the purpose of a screening project is to identify novel lead compounds then it is desirable to screen a collections with chemically diverse structures to increase the likelihood to identify novel scaffolds that may become leads. This is commonly known as sampling a diverse region of chemical space (Medina-Franco, 2012). However, if the purpose of the screening campaign is to optimize one or more specific chemical scaffolds, then it is desirable to explore dense regions of chemical space, e.g. screening combinatorial libraries (Houghten et al., 2008).
There are at least two major approaches to assess the structural diversity. One of them is based on structural fingerprints for example using molecular fragments or pharmacophoric features. The second approach uses chemical scaffolds that are an intuitive way to represent chemical structures (Brown and Jacoby, 2006). The structural diversity of natural products databases either using structural fingerprints and molecular scaffolds have been reported and reviewed in several works (Koch et al., 2005, Ertl et al., 2008, Singh et al., 2009, Chen et al. , 2012a). For example, Yongye and Medina-Franco reported a scaffold-based analysis of five natural products collections whose chemical structures are available in the public domain (Yongye et al. , 2012). The natural products libraries were compared with a general screening collection and natural products libraries frequently used in in vivo screening. It was concluded that the general screening library had the largest scaffold diversity. Other than the benzene and acyclic molecules, flavones, coumarins, and flavanones were identified as the most frequent scaffolds across the various natural products databases (Medina-Franco et al., 2013).
Structural complexity
The concept of structural complexity has broad implications because there is evidence that the increased structural complexity is associated with increased structural specificity. As commented elsewhere (López-Vallejo et al. , 2012) Dandapani and Mar-caurelle pointed out that natural products have larger structural complexity than commercially available compounds as measured by the fraction of saturated carbons (Dandapani and Marcaurelle, 2010). Clemons et al. reported an experimental profile of more than 15000 compounds tested with 100 diverse proteins. The compound libraries included molecules from natural origin and synthetic analogues, commercial compounds and molecules synthesized by academic groups. As a result he concluded that indeed increasing molecular complexity enhance selectivity and frequency of binding (Clemons et al., 2010). In a follow up study Yongye and Medina-Franco reported a chemoinformatic analysis of the Clemons' data set using SPID (Structure-Promiscuity Index Difference), a measure designed to rapidly capture large changes in binding profiles due to small changes in molecular structure. That study revealed that, in general, similar synthetic structures from academic groups showed greater promiscuity differences than do natural products and commercial compounds (Yongye and Medina-Franco, 2012).
In a separate work, it was demonstrated quantitatively the increased structural complexity of natural products in the TCM database (see above) as compared to commercially available databases. In that study it was found that a set of combinatorial libraries that occupy a more dense region in chemical space than TCM (i.e., they have less structural diversity), also have large structural complexity. The major outcome of that analysis was that natural products from TCM along with combinatorial libraries analyzed in that work are good candidate libraries to expand the medicinally relevant chemical space as defined by currently approved drugs (López-Vallejo et al., 2012).
Profile of physicochemical properties
Physicochemical profiling of compound datasets is at the core of currently many empirical rules that try to define drug- and lead-likeness. A prominent example of such empirical rule is the Lipinski's Rule of Five (Lipinski et al., 1997) that has been largely revised over the past recent years (Leeson and Davis, 2004, Faller et al., 2011, López-Vallejo et al., 2012). Several studies have addressed the analysis of the distribution of physicochemical properties of different natural products databases (Feher and Schmidt, 2003, Singh et al., 2009, Medina-Franco et al. , 2012). One of the classical examples is the work of Feher et al. that compared more than 40 molecular properties of natural products assembled from various sources, drugs and compounds obtained from combinatorial chemistry. It was concluded that natural products differ from combinatorial compounds in the number of chiral centers, aromatic rings, number of complex ring systems, degree of saturation of the molecule and the number and ratios of various heteroatoms (Feher and Schmidt, 2003).
More recently, Medina-Franco et al. compared the physicochemical properties of natural products collections with chemical structures freely available with more than 2000 food materials designated as ''Generally Recognized as Safe'' (GRAS) (Medina-Franco et al., 2012). Authors concluded that natural products collections obtained from different sources have different distributions of physicochemical properties and structural diversity in support of previous conclusions derived from the scaffold analysis of the same databases (Yongye et al., 2012). It was also found that the GRAS compounds analyzed in that work have a high structural diversity, comparable to the high structural diversity of natural products and other reference libraries (Medina-Franco et al., 2012).
Manallack et al. carried out an analysis of the distribution of ionization constants of 89425 natural products available in ZINC for purchase and testing (Manallack et al., 2013b). The profile was compared to other screening collections available in ZINC, drugs and a chemogenomics data set. In that study authors found that natural products have different distribution of ionization constants than other screening collections e.g. higher proportions of complex ionizable compounds and a greater number of zwitterionic molecules. However, natural products from ZINC have some overlap with approved drugs. The distribution of pKa values of single acids and single bases in natural products were more similar to drugs than screening compounds (Manallack et al. , 2013b). In a follow-up work Manallack et al. performed a similar characterization of the acid/base profile of 25566 natural products obtained from ChEMBL (Manallack et al., 2013a). In the later study, the profile was compared with human small molecule metabolites.
Visualization of the chemical space
Chemical space has several definitions. For example, Dobson defines the chemical space as 'the total descriptor space that encompasses all the small carbon-based molecules that could in principle be created' (Dobson, 2004). In a more intuitive concept of space Lipinski and Hopkins mention that 'chemical space can be viewed as being analogous to the cosmological universe in its vastness, with chemical compounds populating space instead of stars' (Lipinski and Hopkins, 2004).
The concept of chemical space has a major role in drug discovery projects since it helps to classify and compare compound data sets. This concept is also commonly used in library design and compound selection for experimental testing (Reymond et al., 2010, Medina-Franco et al., 2008). Data visualization has an important role to rapidly mine the constantly increasing information available for drug discovery. There are several established methods to visualize the chemical space (Medina-Franco et al., 2008) and recent advances in the visualization of chemogenomics data sets have been reviewed (Medina-Franco and Aguayo-Ortiz, 2013).
A visual representation of the chemical space covered by TCM was reported using principal component analysis of six druglike physicochemical properties (López-Vallejo et al., 2012). It was clear in that work that TCM occupy a different region of chemical space occupied by currently approved drugs. In a separate analysis, the chemical space of natural products from a commercial vendor was visualized with the space covered by GRAS compounds using self-organizing-maps (Medina-Franco et al., 2012).
Gu et al. generated a visual representation of the chemical space covered by the UNPD (discussed above) with FDA-approved drugs. A three-dimensional representation of the chemical space was generated using principal component analysis of 15 molecular descriptors (Gu et al., 2013). This representation clearly showed the large diversity of UNPD and the overlap of this database with the chemical space of drugs.
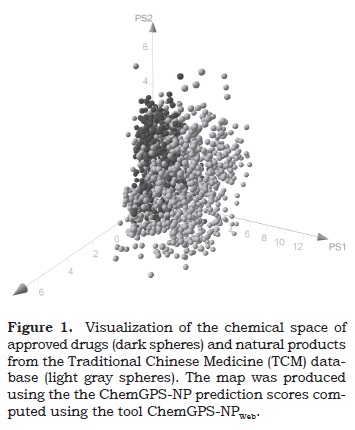
To illustrate the visualization of chemical space, Figure 1 shows a visual representation of the chemical space of 1000 approved drugs obtained from DrugBank database (http://www.drugbank.ca/) (dark spheres) and 997 compounds randomly selected from the TCM database using molecular properties (light gray spheres). The visualization was generated using the online tool ChemGPS-NPWeb (Larsson et al., 2007, Rosen et al., 2009). ChemGPS-NP (Larsson et al., 2005, Larsson et al., 2007) is a global chemical positioning system based on principal component analysis (PCA) (Oprea and Gottfries, 2001) and it is suited for exploration of biologically relevant chemical space. The first four dimensions of the ChemGPS-NP plot preserve 77% of data variance. The first dimension (PC1) represents size, shape and polarizability (main contribution is size); PC2 is associated with aromatic and conjugation related properties (main influence is aromaticity); PC3 describes lipophilicity, polarity, and H-bond capacity (major contribution is lipophilicity); and PC4 expresses flexibility and rigidity. Chemical compounds can be positioned onto this map using interpolation using PCA score prediction. More details of this approach as described elsewhere (Rosen et al., 2009). Figure 1 shows that TCM and drugs have a partial overlap in the property space. The figure also shows the large property diversity of TCM. ChemGPS-NPWeb has recently been used to compare the chemical space of drugs with in-house combinatorial libraries (Medina-Franco and Waddell, 2012).
MOLECULAR MODELING
There are numerous molecular modeling studies focused on the elucidation of the activity of bioactive natural products, e.g., docking of individual compounds with a particular molecular target. An illustrative example is the analysis of the binding mode of curcumin, pathenolide, and (-)-epigallocathechin-3-gallate with the enzyme DNA methyltransferase (DNMT), a major epigenetic target for the treatment of cancer and other diseases (Yoo and Medina-Franco, 2011, Yoo and Medina-Franco, 2012). Structure-based pharma-cophore modeling and docking of natural products have been extensively used to identify key protein-ligand interactions that can be used for the optimization of DNMT inhibitors. Other molecular modeling studies of active natural products with diverse molecular targets are continuously performed (El-Elimat et al., 2013, Ramírez-Espinosa et al., 2013). Geldenhuys et al. recently reviewed the role of resveratrol, curcumin, caffeine, and genestein as starting compounds for molecular modeling studies (Geldenhuys et al., 2012).
In the following sub-sections we focus the application of molecular modeling to identify compounds from natural origin for a target of interest (using virtual screening) and elucidate potential molecular targets of natural products (using target fishing). Virtual screening and target fishing are two broad techniques that have been recently discussed in an integrated manner with other concepts relevant to chemogenomics (Medina-Franco et al., 2013).
Virtual screening
Since systematic experimental screening of large chemical databases is a time consuming and expensive process, computational (in silico or virtual) screening is used as means to filter compound databases to select compounds for biological testing (Scior et al., 2012). The type of libraries that can be screened are collections of molecules physically available for experimental testing or virtual libraries. In the second case, the computational hits are selected for synthesis and then testing. Ideally, virtual screening should be part of an iterative process that involves the prediction, experimental testing of selected compounds and design of new chemical data sets based on the structure of the experimental hits. The new molecules can be designed using a combination of medicinal chemistry and computational tools. Although it is highly desirable to identify potent compounds in the first round of virtual screening and experimental testing, the major goal of the initial screening is to identify hit compounds with novel chemical scaffolds for later optimization (Muegge, 2008). The method or series of approaches to conduct virtual screening can be divided in two major groups, structure-based and ligand-based, depending on the experimental information available. Ligand-based approaches utilize structure-activity data of a set of known actives. Structure-based methods use the three-dimensional structure of the biological target. Whenever possible, it is recommended to use a combination of ligand- and structure-based methods. The interested reader is directed to extensive reviews of virtual screening that include methods, successful applications, pitfalls and workarounds (Shoichet, 2004, Villoutreix et al., 2009, Ripphausen et al., 2011, López-Vallejo et al., 2011, Guido et al., 2008, Muegge, 2008, Scior et al., 2012). Advances in the virtual screening of virtual compounds have been reviewed by Reymond et al. (Reymond and Awale, 2012). Ma et al. have reviewed the synergy between structure-based virtual screening and drug repurposing (Ma et al., 2013).
A recent example of successful virtual screening using natural product databases is exemplified by the work of Cao et al. (Cao et al., 2013). In that work, the authors conducted virtual screening of an in-house collection with more than 4000 natural products isolated from 100 medicinal plants. The collection was docked into the ligand binding domain of estrogen receptors (ER) ERα and ERβ using the program Glide. As a result, eleven natural non-steroidal ER modulators were identified (Cao et al., 2013).
Guasch et al. identified five peroxisome proliferator-activated receptors, PPARy partial agonists from a pool of more than 89000 natural products and natural products derivatives from ZINC (Guasch et al., 2012). In that work, the initial database was filtered sequentially using ADMETox filters, structure-based pharmacophore screening, molecular docking, electrostatic, and fingerprint-based similarity analysis. Ten compounds were selected for in vitro validation assays and half of the molecules showed activity (Guasch et al., 2012).
There are comprehensive virtual screening studies of compound databases that strongly suggest activity of natural products as lead compounds. In one study, Medina-Franco et al. conducted the docking-based virtual screening of a lead-like set of natural products with a validated homology model of the catalytic domain of human DNMT1 (Medina-Franco et al., 2011). A multi-step docking approach was implemented using first fast docking-based protocols with Glide high-throughput virtual screening and Glide Standard Precision. Then, a parallel docking protocol was implemented using three different docking programs, namely Glide Extra Precision, Genetic Optimization for Ligand Docking (GOLD) and Autodock. Consensus hits e.g., top ranked compound by all three docking programs, were selected as promising candidates for experimental testing. Interestingly, one of the computational hits was already experimentally reported to be an actual inhibitor (Kuck et al., 2010), thus validating the computational approach (Medina-Franco et al., 2011).
A second example of a recent study is given by the work of Ngo et al. that predicted the binding affinity of 342 molecules obtained from Vietnamese plants to the full-length amyloid Aβ1-40 and Aβ1-42 peptides and their mature fibrils. The authors used a combination of docking using the program Autodock Vina, and molecular dynamics. It was concluded that five natural are promising candidates for the development of molecules to treat Alzheimer's disease (Ngo and Li, 2013).
As illustrated above, many researchers use in-house, commercial or non-commercial computational tools to screen existing natural products databases with a molecular target of interest. Particularly attractive for experimental; groups an alternative to conduct computational screening of compound databases is to establish collaborations with expert computational groups to conduct the screening. In natural products research a remarkable example is the Drug Discovery Portal (DDP). This initiative is hosted at the University of Strathclyde and it is intended to improve drug discovery collaborations between academic groups of chemists and biologists (Clark et al., 2010). At the same time, DDP seek to preserve the intellectual property rights. This initiative provides the service to conduct the in-silico screening of collections available at DDP. In addition, DDP can provide to the collaborator with physical samples of the computational hits for experimental testing.
Target fishing and reverse pharmacognosy
As reviewed above, virtual screening aims to identify new ligands for known targets. The inverse approach i.e., identify putative targets for known ligands, is referred in the literature as target fishing (Rognan, 2010). Thus, virtual screening is related to ligand screening and target fishing is associated with ligand profiling (Jacoby, 2011). Similar to virtual screening, depending on the experimental information available, target fishing can be performed using structure-based methods e.g., inverse docking, or ligand-based methods e.g., similarity searching (Nettles et al., 2006, AbdulHameed et al., 2011) or both.
Yue et al. have reviewed advances in the target profiling of natural products using experimental (genomics and proteomics) and computational approaches (Yue et al. , 2012). In that rich review, authors emphasize the need of integrating various techniques including docking compounds across different targets (inverse docking), mapping ligand-target profiling space, and network analysis (Yue et al., 2012).
Drug-target interaction networks is at the core of target fishing as it is an approach that has been successfully used to predict putative targets based on the chemical similarity of the known ligands (Keiser et al., 2009, Besnard et al., 2012, Cheng et al. , 2013). In a short but insightful review Gertsch highlights the relevance of analyzing ligand-target networks for botanical drugs (Gertsch, 2011).
Gu et al. (Gu et al., 2011) conducted the virtual screening of 676 compounds from the TCM database with 37 proteins related to type II diabetes mellitus. Interaction networks were used to link compounds with proteins based on the docking score results. Authors elucidated the action mechanism of a medical composition which had clinical efficacy for type II diabetes mellitus (Gu et al. , 2011). More recently, the same authors dock the UNPD (see above) with 332 molecular targets of FDA-approved drugs (Gu et al. , 2013). Based on the docking scores and the natural product-target networks the most promising natural products for drug discovery in UNPD were selected (Gu et al., 2013).
As part of the FoodInformatics symposia held at the 245th American Chemical Society National Meeting in 2013 (Martínez-Mayorga et al., 2013), Quoc-Tuan Do explained the principles of the concept reverse pharmacognosy (Blondeau et al., 2010) emphasizing the many roles of chemoinformatic approaches, including inverse screening, to accelerate the identification of the bioactive compounds of an organism. During the symposia Quoc-Tuan discussed two successful and published examples of reverse pharmacognosy using SelnergyTM, a platform developed in Greenpharma to predict, based on docking, interaction energies of a ligand with a target protein (Do et al., 2007, Bernard et al., 2008).
CONCLUSIONS
Natural products have been a major component in drug discovery providing, for many years, lead compounds approved for clinical use and inspiring the synthesis of chemical libraries. The synergy of experimental natural products research with computational approaches is increasing. Chemoinformatics methods have been able to characterize the chemical space of public and commercially available natural products databases comparing their molecular properties, including structural complexity, and structural diversity with approved drugs and other screening libraries. The number of natural products databases publicly available is increasing. These databases are being assembled in several different countries and regions including Latin America and Asia. Molecular modeling is continuously applied to suggest binding models of bioactive natural products with their molecular targets. This has been helpful to further understand, at the molecular level, the biological activity of these compounds and to guide the chemical synthesis of natural products analogues with improved activity. Virtual screening has been used to conduct the systematic search of bioactive natural products with a molecular target of interest. Conversely, target fishing approaches have also been employed to identify potential molecular targets of natural products. It is anticipated the continued synergy of experimental natural products research with compute-raided drug design to further advance drug discovery.
ACKNOWLEDGEMENTS
We thank Jacob Waddell his help to prepare figure 1. This work was supported by the State of Florida.
REFERENCES
AbdulHameed, M. D. M., Chaudhury, S., Singh, N., Sun, H., Wallqvist, A. & Tawa, G. J. 2011. Exploring polypharmacology using a ROCS-based target fishing approach. Journal of Chemical Information and Modeling 52: 492-505. [ Links ]
Barbosa, A. J. M. & Del Rio, A. 2012. Freely accessible databases of commercial compounds for high- throughput virtual screenings. Current Topics in Medicinal Chemistry 12: 866-877. [ Links ]
Barlow, D. J., Buriani, A., Ehrman, T., Bosisio, E., Eberini, I. & Hylands, P. J. 2012. In-silico studies in Chinese herbal medicines' research: Evaluation of in-silico methodologies and phytochemical data sources, and a review of research to date. Journal of Ethnopharmacology 140: 526-534. [ Links ]
Bernard, P., Dufresne-Favetta, C., Favetta, P., Do, Q. T., Himbert, F., Zubrzycki, S., Scior, T. & Lugnier, C. 2008. Application of drug repositioning strategy to TOFISOPAM. Current Medicinal Chemistry 15: 3196-3203. [ Links ]
Besnard, J., Ruda, G. F., Setola, V., Abecassis, K., Rodriguiz, R. M., Huang, X. P., Norval, S., Sassano, M. F., Shin, A. I., Webster, L. A., Simeons, F. R. C., Stojanovski, L., Prat, A., Seidah, N. G., Constam, D. B., Bickerton, G. R., Read, K. D., Wetsel, W. C., Gilbert, I. H., Roth, B. L. & Hopkins, A. L. 2012. Automated design of ligands to polypharmacological profiles. Nature 492: 215-220. [ Links ]
Bienstock, R. J. 2012. Computational drug design targeting protein-protein interactions. Current Pharmaceutical Design 18: 1240-1254. [ Links ]
Blondeau, S., Do, Q. T., Scior, T., Bernard, P. & Morin-Allory, L. 2010. Reverse pharmacognosy: Another way to harness the generosity of Nature. Current Pharmaceutical Design 16: 1682-1696. [ Links ]
Bohlin, L., Göransson, U., Alsmark, C., Wedén, C. & Backlund, A. 2010. Natural products in modern life science. Phytochemistry Reviews 9: 279-301. [ Links ]
Brown, N. & Jacoby, E. 2006. On scaffolds and hopping in medicinal chemistry. Mini-Revies in Medicinal Chemistry 6: 1217-1229. [ Links ]
Cao, X., Jiang, J., Zhang, S., Zhu, L., Zou, J., Diao, Y., Xiao, W., Shan, L., Sun, H., Zhang, W., Huang, J. & Li, H. 2013. Discovery of natural estrogen receptor modulators with structure-based virtual screening. Bioorganic & Medicinal Chemistry Letters 23: 3329-33. [ Links ]
Chen, C. Y.-C. 2011. TCM Database@Taiwan: The World's largest traditional chinese medicine database for drug screening in silico. PLoS ONE 6. [ Links ]
Chen, H., Engkvist, O., Blomberg, N. & Li, J. 2012a. A comparative analysis of the molecular topologies for drugs, clinical candidates, natural products, human metabolites and general bioactive compounds. MedChemComm 3: 312-321. [ Links ]
Chen, K.-Y., Chang, S.-S. & Chen, C. Y.-C. 2012b. In Silico identification of potent pancreatic triacylglycerol lipase inhibitors from Traditional Chinese Medicine. PLoS ONE 7: e43932. [ Links ]
Chen, L., Morrow, J. K., Tran, H. T., Phatak, S. S., Du-Cuny, L. & Zhang, S. X. 2012c. From laptop to benchtop to bedside: Structure-based drug design on protein targets. Current Pharmaceutical Design 18: 1217-1239. [ Links ]
Cheng, F., Li, W., Wu, Z., Wang, X., Zhang, C., Li, J., Liu, G. & Tang, Y. 2013. Prediction of polypharmacological profiles of drugs by the integration of chemical, side effect, and therapeutic space. Journal of Chemical Information and Modeling 53: 753-62. [ Links ]
Clark, R. L., Johnston, B. F., Mackay, S. P., Breslin, C. J., Robertson, M. N. & Harvey, A. L. 2010. The Drug Discovery Portal: a resource to enhance drug discovery from academia. Drug Discovery Today 15: 679-683. [ Links ]
Clemons, P. A., Bodycombe, N. E., Carrinski, H. A., Wilson, J. A., Shamji, A. F., Wagner, B. K., Koehler, A. N. & Schreiber, S. L. 2010. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proceedings ofthe National Academy of Sciences USA 107: 18787-18792. [ Links ]
Dandapani, S. & Marcaurelle, L. A. 2010. Accessing new chemical space for 'undruggable' targets. Nature Chemical Biology 6: 861-863. [ Links ]
Do, Q. T., Lamy, C., Renimel, I., Sauvan, N., Andre, P., Himbert, F., Morin-Allory, L. & Bernard, P. 2007. Reverse pharmacognosy: Identifying biological 3 properties for plants by means of their molecule constituents: Application to meranzin. Planta Medica 73: 1235-1240. [ Links ]
Dobson, C. M. 2004. Chemical space and biology. Nature 432: 824-828. [ Links ]
Duffy, B. C., Zhu, L., Decornez, H. & Kitchen, D. B. 2012. Early phase drug discovery: Cheminformatics and computational techniques in identifying lead series. Bioorganic & Medicinal Chemistry 20: 5324-5342. [ Links ]
Durrant, J. & McCammon, J. A. 2011. Molecular dynamics simulations and drug discovery. BMC Biology 9: 71. [ Links ]
El-Elimat, T., Figueroa, M., Raja, H. A., Graf, T. N., Adcock, A. F., Kroll, D. J., Day, C. S., Wani, M. C., Pearce, C. J. & Oberlies, N. H. 2013. Benzoquinones and terphenyl compounds as phosphodiesterase-4B inhibitors from a fungus of the order chaetothyriales (MSX 47445). Journal of Natural Products 76: 382-387. [ Links ]
Engel, T. 2006. Basic overview of chemoinformatics. Journal of Chemical Information and Modeling 46: 2267-2277. [ Links ]
Ertl, P., Roggo, S. & Schuffenhauer, A. 2008. Natural product-likeness score and its application for prioritization of compound libraries. Journal of Chemical Information and Modeling 48: 68-74. [ Links ]
Faller, B., Ottaviani, G., Ertl, P., Berellini, G. & Collis, A. 2011. Evolution of the physicochemical properties of marketed drugs: can history foretell the future? Drug Discovery Today 16: 976-984. [ Links ]
Feher, M. & Schmidt, J. M. 2003. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. Journal of Chemical Information and Computer Sciences 43: 218-227. [ Links ]
Ganesan, A. 2008. The impact of natural products upon modern drug discovery. Current Opinion in Chemical Biology 12: 306-317. [ Links ]
Geldenhuys, W. J., Bishayee, A., Darvesh, A. S. & Carroll, R. T. 2012. Natural products of dietary origin as lead compounds in virtual screening and drug design. Current Pharmaceutical Biotechnology 13: 117-124. [ Links ]
Gertsch, J. 2011. Botanical drugs, synergy, and network pharmacology: Forth and back to intelligent mixtures. Planta Medica 77: 1086-1098. [ Links ]
Gu, J., Gui, Y., Chen, L., Yuan, G., Lu, H.-Z. & Xu, X. 2013. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE 8: e62839. [ Links ]
Gu, J., Zhang, H., Chen, L., Xu, S., Yuan, G. & Xu, X. 2011. Drug-target network and po-lypharmacology studies of a Traditional Chinese Medicine for type II diabetes mellitus. Computational Biology and Chemistry 35: 293-297. [ Links ]
Guasch, L., Sala, E., Castell-Auvi, A., Cedo, L., Liedl, K. R., Wolber, G., Muehlbacher, M., Mulero, M., Pinent, M., Ardevol, A., Valls, C., Pujadas, G. & Garcia-Vallve, S. 2012. Identification of PPARgamma partial agonists of natural origin (I): Development of a virtual screening procedure and in vitro Validation. PLoS ONE 7: e50816 [ Links ]
Guido, R. V. C., Oliva, G. & Andricopulo, A. D. 2008. Virtual screening and its integration with modern drug design technologies. Current Medicinal Chemistry 15: 37-46. [ Links ]
Harvey, A. L. 2008. Natural products in drug discovery. Drug Discovery Today 13: 894-901. [ Links ]
Houghten, R. A., Pinilla, C., Giulianotti, M. A., Appel, J. R., Dooley, C. T., Nefzi, A., Ostresh, J. M., Yu, Y. P., Maggiora, G. M., Medina-Franco, J. L., Brunner, D. & Schneider, J. 2008. Strategies for the use of mixture-based synthetic combinatorial libraries: Scaffold ranking, direct testing, in vivo, and enhanced deconvolution by computational methods. Journal of Combinatorial Chemistry 10: 3-19. [ Links ]
Irwin, J. J. & Shoichet, B. K. 2005. ZINC - A free database of commercially available compounds for virtual screening. Journal of Chemical Information and Modeling 45: 177-182. [ Links ]
Jacoby, E. 2011. Computational chemogenomics. Wiley Interdisciplinary Reviews: Computational Molecular Science 1 : 57-67. [ Links ]
Keiser, M. J., Setola, V., Irwin, J. J., Laggner, C., Abbas, A. I., Hufeisen, S. J., Jensen, N. H., Kuijer, M. B., Matos, R. C., Tran, T. B., Whaley, R., Glennon, R. A., Hert, J., Thomas, K. L. H., Edwards, D. D., Shoichet, B. K. & Roth, B. L. 2009. Predicting new molecular targets for known drugs. Nature 462: 175-U48. [ Links ]
Koch, M. A., Schuffenhauer, A., Scheck, M., Wetzel, S., Casaulta, M., Odermatt, A., Ertl, P. & Waldmann, H. 2005. Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proceedings of the National Academy of Sciences USA 102: 17272-17277. [ Links ]
Kuck, D., Singh, N., Lyko, F. & Medina-Franco, J. L. 2010. Novel and selective DNA methyltransferase inhibitors: Docking-based virtual screening and experimental evaluation. Bioorganic & Medicinal Chemistry 18: 822-829. [ Links ]
Lachance, H., Wetzel, S., Kumar, K. & Waldmann, H. 2012. Charting, navigating, and populating natural product chemical space for drug discovery. Journal of Medicinal Chemistry 55: 5989-6001. [ Links ]
Larsson, J., Gottfries, J., Bohlin, L. & Backlund, A. 2005. Expanding the ChemGPS chemical space with natural products. Journal of Natural Products 68: 985-991. [ Links ]
Larsson, J., Gottfries, J., Muresan, S. & Backlund, A. 2007. ChemGPS-NP: Tuned for navigation in biologically relevant Chemical space. Journal of Natural Products 70: 789-794. [ Links ]
Leeson, P. D. & Davis, A. M. 2004. Time-related differences in the physical property profiles of oral drugs. Journal of Medicinal Chemistry 47: 6338-6348. [ Links ]
Li, J. W.-H. & Vederas, J. C. 2009. Drug discovery and natural products: End of an era or an endless frontier? Science 325: 161-165. [ Links ]
Liao, C. Z., Sitzmann, M., Pugliese, A. & Nicklaus, M. C. 2011. Software and resources for computational medicinal chemistry. Future Medicinal Chemistry 3: 1057-1085. [ Links ]
Lipinski, C. & Hopkins, A. 2004. Navigating chemical space for biology and medicine. Nature 432: 855-861. [ Links ]
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. 1997. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 23: 3-25. [ Links ]
López-Vallejo, F., Caulfield, T., Martínez-Mayorga, K., Giulianotti, M. A., Nefzi, A., Houghten, R. A. & Medina-Franco, J. L. 2011. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Combinatorial Chemistry & High Throughput Screening 14: 475-487. [ Links ]
López-Vallejo, F., Giulianotti, M. A., Houghten, R. A. & Medina-Franco, J. L. 2012. Expanding the medicinally relevant chemical space with compound libraries. Drug Discovery Today 17: 718-726. [ Links ]
Ma, D.-L., Chan, D. S.-H. & Leung, C.-H. 2013. Drug repositioning by structure-based virtual screening. Chemical Society Reviews 42: 2130-2141. [ Links ]
Manallack, D. T., Dennis, M. L., Kelly, M. R., Prankerd, R. J., Yuriev, E. & Chalmers, D. K. 2013a. The Acid/Base profile of the human metabolome and natural products. Molecular Informatics, in press. DOI: 10.1002/minf.201200167. [ Links ]
Manallack, D. T., Prankerd, R. J., Nassta, G. C., Ursu, O., Oprea, T. I. & Chalmers, D. K. 2013b. A Chemogenomic analysis of ionization constants-Implications for drug discovery. ChemMedChem 8: 242-255. [ Links ]
Martínez-Mayorga, K., Medina-Franco, J. L. & Organizers 2013. FoodInformatics: Applications of Chemical Information to Food Chemistry. Division of Chemical Information. 245th ACS National Meeting, New Orleans, LI, United States. New Orleans, LI, United States: American Chemical Society, Washington, D. C. [ Links ]
Medina-Franco, J. L. 2012. Interrogating novel areas of chemical space for drug discovery using chemoinformatics. Drug Development Research 73: 430-438. [ Links ]
Medina-Franco, J. L. 2013 Chemoinformatics characterization of the chemical space and molecular diversity of compound libraries. In Andrea, T. (ed) Diversity-oriented synthesis:Basics and applications in organic synthesis, drug discovery, and chemical biology. John Wiley & Sons, Inc. p. 325-352. [ Links ]
Medina-Franco, J. L. & Aguayo-Ortiz, R. 2013. Progress in the visualization and mining of chemical and target spaces. Molecular Informatics, in press. [ Links ]
Medina-Franco, J. L., Giulianotti, M. A., Welmaker, G. S. & Houghten, R. A. 2013. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discovery Today 18: 495-501. [ Links ]
Medina-Franco, J. L., Lopez-Vallejo, F. & Castillo, R. 2006. Diseño de fármacos asistido por computadora. Educación Química 17: 452-457. [ Links ]
Medina-Franco, J. L., López-Vallejo, F., Kuck, D. & Lyko, F. 2011. Natural products as DNA methyltransferase inhibitors: a computer-aided discovery approach. Molecular Diversity 15: 293-304. [ Links ]
Medina-Franco, J. L., Martínez-Mayorga, K., Giulianotti, M. A., Houghten, R. A. & Pinilla, C. 2008. Visualization of the chemical space in drug discovery. Current Computer-Aided Drug Design 4: 322-333. [ Links ]
Medina-Franco, J. L., Martínez-Mayorga, K., Peppard, T. L. & Del Rio, A. 2012. Chemoinformatic analysis of GRAS (Generally Recognized as Safe) flavor chemicals and natural products. PLoS ONE 7: e50798. [ Links ]
Medina-Franco, J. L. & Waddell, J. 2012. Towards the bioassay activity landscape modeling in compound databases. Journal of the Mexican Chemical Society 56: 163-168. [ Links ]
Muegge, I. 2008. Synergies of virtual screening approaches. Mini-Reviews in Medicinal Chemistry 8: 927-933. [ Links ]
Nettles, J. H., Jenkins, J. L., Bender, A., Deng, Z., Davies, J. W. & Glick, M. 2006. Bridging chemical and biological space: "Target fishing" using 2D and 3D molecular descriptors. Journal of Medicinal Chemistry 49: 6802-6810. [ Links ]
Newman, D. J. 2008. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? Journal of Medicinal Chemistry 51 : 2589-2599. [ Links ]
Ngo, S. T. & Li, M. S. 2013. Top-leads from natural products for treatment of Alzheimer's disease: docking and molecular dynamics study. Molecular Simulation 39: 279-291. [ Links ]
Nicola, G., Liu, T. & Gilson, M. K. 2012. Public domain databases for medicinal chemistry. Journal of Medicinal Chemistry 55: 6987-7002. [ Links ]
Oprea, T. I. & Gottfries, J. 2001. Chemography: The art of navigating in chemical space. Journal of Combinatorial Chemistry 3: 157-166. [ Links ]
Ou-Yang, S.-s., Lu, J.-y., Kong, X.-q., Liang, Z.-j., Luo, C. & Jiang, H. 2012. Computational drug discovery. Acta Pharmacologica Sinica 33: 1131-1140. [ Links ]
Ramírez-Espinosa, J. J., García-Jiménez, S., Rios, M. Y., Medina-Franco, J. L., López-Vallejo, F., Webster, S. P., Binnie, M., Ibarra-Barajas, M., Ortiz-Andrade, R. & Estrada-Soto, S. 2013. Antihyperglycemic and sub-chronic antidiabetic actions of morolic and moronic acids, in vitro and in silico inhibition of 11p-HSD 1. Phytomedicine 20: 571-576. [ Links ]
Reymond, J.-L. & Awale, M. 2012. Exploring chemical space for drug discovery using the Chemical Universe Database. ACS Chemical Neuroscience 3: 649-657. [ Links ]
Reymond, J.-L., van Deursen, R., Blum, L. C. & Ruddigkeit, L. 2010. Chemical space as a source for new drugs. MedChemComm 1 : 30-38. [ Links ]
Ripphausen, P., Nisius, B. & Bajorath, J. 2011. State-of-the-art in ligand-based virtual screening. Drug Discovery Today 16: 372-376. [ Links ]
Rognan, D. 2010. Structure-Based Approaches to target fishing and ligand profiling. Molecular Informatics 29: 176-187. [ Links ]
Rosen, J., Lovgren, A., Kogej, T., Muresan, S., Gottfries, J. & Backlund, A. 2009. ChemGPS-NPWeb: chemical space navigation online. Journal of Computer-Aided Molecular Design 23: 253-259. [ Links ]
Sanders, M. P. A., Barbosa, A. J. M., Zarzycka, B., Nicolaes, G. A. F., Klomp, J. P. G., de Vlieg, J. & Del Rio, A. 2012. Comparative analysis of pharmacophore screening tools. Journal of Chemical Information and Modeling 52: 1607-1620. [ Links ]
Scior, T., Bender, A., Tresadern, G., Medina-Franco, J. L., Martínez-Mayorga, K., Langer, T., Cuanalo-Contreras, K. & Agrafiotis, D. K. 2012. Recognizing pitfalls in virtual screening: A critical review. Journal of Chemical Information and Modeling 52: 867-881. [ Links ]
Scior, T., Bernard, P., Medina-Franco, J. L. & Maggiora, G. M. 2007. Large compound databases for structure-activity relationships studies in drug discovery. Mini-Reviews in Medicinal Chemistry 7: 851-860. [ Links ]
Shoichet, B. K. 2004. Virtual screening of chemical libraries. Nature 432: 862-865. [ Links ]
Singh, N., Guha, R., Giulianotti, M. A., Pinilla, C., Houghten, R. A. & Medina-Franco, J. L. 2009. Chemoinformatic analysis of combinatorial libraries, drugs, natural products, and molecular libraries small molecule repository. Journal of Chemical Information and Modeling 49: 1010-1024. [ Links ]
Sinko, W., Lindert, S. & McCammon, J. A. 2013. Accounting for receptor flexibility and enhanced sampling methods in computer-aided drug design. Chemical Biology & Drug Design 81: 41-49. [ Links ]
Tsai, T.-Y., Chang, K.-W. & Chen, C. 2011. iScreen: world's first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. Journal of Computer-Aided Molecular Design 25: 525-531. [ Links ]
Valli, M., dos Santos, R. N., Figueira, L. D., Nakajima, C. H., Castro-Gamboa, I., Andricopulo, A. D. & Bolzani, V. S. 2013. Development of a natural products database from the biodiversity of Brazil. Journal of Natural Products 76: 439-444. [ Links ]
Varnek, A. & Baskin, II 2011. Chemoinformatics as a theoretical chemistry discipline. Molecular Informatics 30: 20-32. [ Links ]
Villoutreix, B. O., Eudes, R. & Miteva, M. A. 2009. Structure-Based virtual ligand screening: Recent success stories. Combinatorial Chemsitry & High Throughput Screening 12: 1000-1016. [ Links ]
Willett, P. 2011. Chemoinformatics: a history. Wiley Interdisciplinary Reviews: Computational Molecular Science 1 : 46-56. [ Links ]
Xiang, M. L., Cao, Y., Fan, W. J., Chen, L. J. & Mo, Y. R. 2012. Computer-aided drug design: Lead discovery and optimization. Combinatorial Chemistry & High Throughput Screening 15: 328-337. [ Links ]
Yongye, A. B. & Medina-Franco, J. L. 2012. Data mining of protein-binding profiling data identifies structural modifications that distinguish selective and promiscuous compounds. Journal of Chemical Information and Modeling 52: 2454-2461. [ Links ]
Yongye, A. B., Waddell, J. & Medina-Franco, J. L. 2012. Molecular scaffold analysis of natural products databases in the public domain. Chemical Biology & Drug Design 80: 717-724. [ Links ]
Yoo, J. & Medina-Franco, J. L. 2011. Homology modeling, docking, and structure-based phar-macophore of inhibitors of DNA methyltransferase. Journal of Computer-Aided Molecular Design 25: 555-567. [ Links ]
Yoo, J. & Medina-Franco, J. L. 2012. Inhibitors of DNA methyltransferases: Insights from computational studies. Current Medicinal Chemistry 19: 3475-3487. [ Links ]
Yue, R., Shan, L., Yang, X. & Zhang, W. 2012. Approaches to target profiling of natural products. Current Medicinal Chemistry 19: 3841-3855. [ Links ]
Yuriev, E. & Ramsland, P. A. 2013. Latest developments in molecular docking: 2010-2011 in review. Journal of Molecular Recognition 26: 215-239. [ Links ]

